

Regulation of hepatic glucose metabolism in health and disease
- PMID: 28731034
- PMCID: PMC5777172
- DOI: 10.1038/nrendo.2017.80
Regulation of hepatic glucose metabolism in health and disease
Abstract
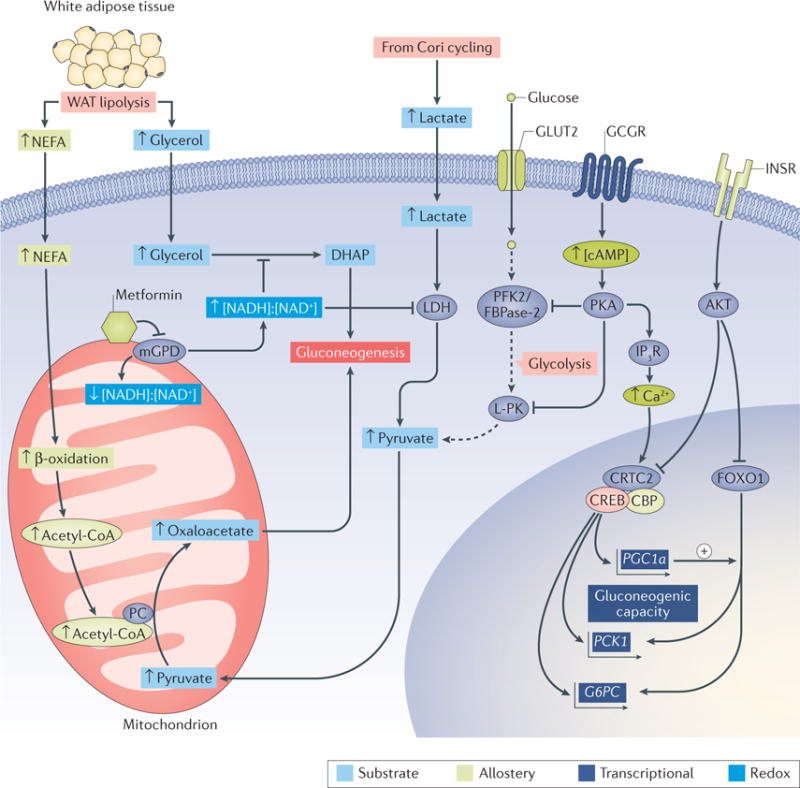
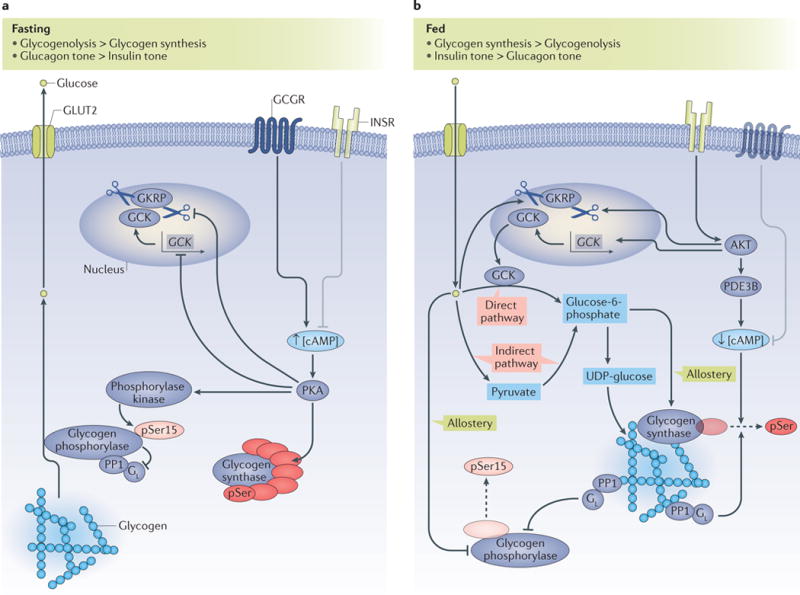
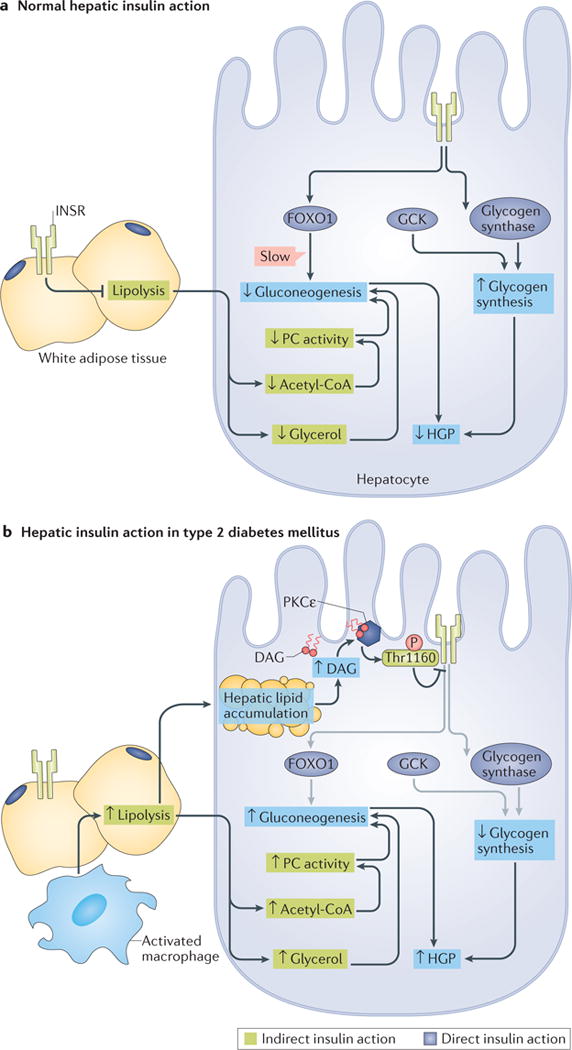
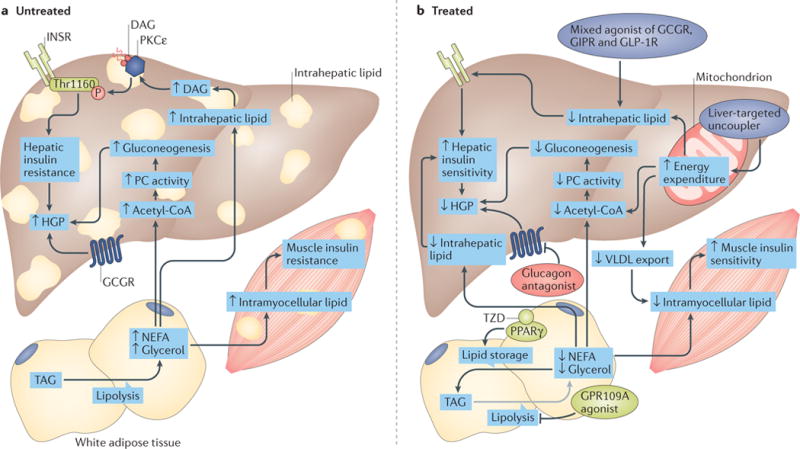
The liver is crucial for the maintenance of normal glucose homeostasis - it produces glucose during fasting and stores glucose postprandially. However, these hepatic processes are dysregulated in type 1 and type 2 diabetes mellitus, and this imbalance contributes to hyperglycaemia in the fasted and postprandial states. Net hepatic glucose production is the summation of glucose fluxes from gluconeogenesis, glycogenolysis, glycogen synthesis, glycolysis and other pathways. In this Review, we discuss the in vivo regulation of these hepatic glucose fluxes. In particular, we highlight the importance of indirect (extrahepatic) control of hepatic gluconeogenesis and direct (hepatic) control of hepatic glycogen metabolism. We also propose a mechanism for the progression of subclinical hepatic insulin resistance to overt fasting hyperglycaemia in type 2 diabetes mellitus. Insights into the control of hepatic gluconeogenesis by metformin and insulin and into the role of lipid-induced hepatic insulin resistance in modifying gluconeogenic and net hepatic glycogen synthetic flux are also discussed. Finally, we consider the therapeutic potential of strategies that target hepatosteatosis, hyperglucagonaemia and adipose lipolysis.
Conflict of interest statement
M.C.P. and D.F.V. declare no competing interests. G.I.S. serves on scientific advisory boards for Merck, Novo Nordisk, Celgene, Aegerion and AstraZeneca, receives investigator-initiated support from Gilead Sciences, Inc., and is an inventor on Yale patents for liver-targeted mitochondrial uncoupling agents for the treatment of NAFLD, NASH, type 2 diabetes and related metabolic disorders.
Figures
References
-
- Ekberg K, et al. Contributions by kidney and liver to glucose production in the postabsorptive state and after 60 h of fasting. Diabetes. 1999;48:292–298. - PubMed
-
- Rothman DL, Magnusson I, Katz LD, Shulman RG, Shulman GI. Quantitation of hepatic glycogenolysis and gluconeogenesis in fasting humans with 13C NMR. Science. 1991;254:573–576. - PubMed
