

ERK/MAPK Signaling Is Required for Pathway-Specific Striatal Motor Functions
- PMID: 28733355
- PMCID: PMC5566864
- DOI: 10.1523/JNEUROSCI.0473-17.2017
ERK/MAPK Signaling Is Required for Pathway-Specific Striatal Motor Functions
Abstract
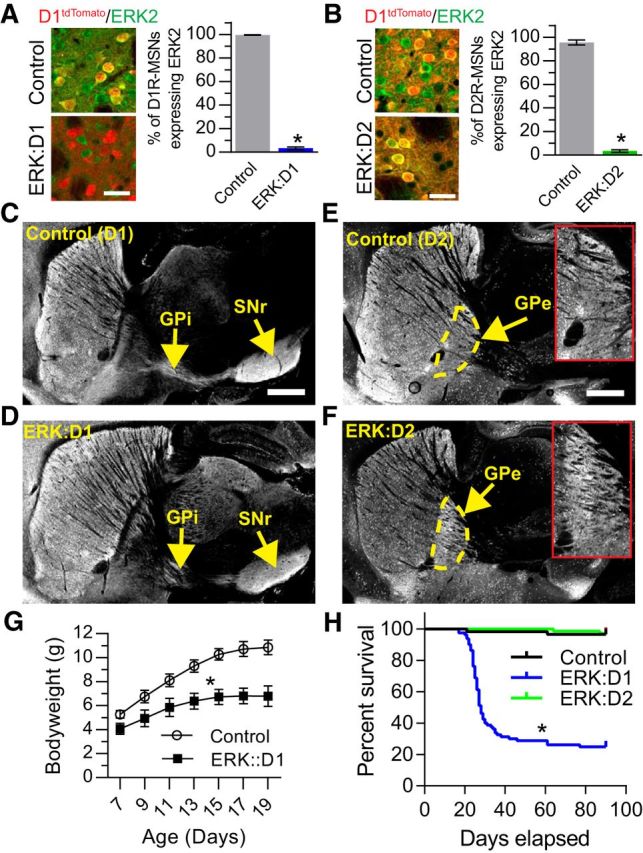
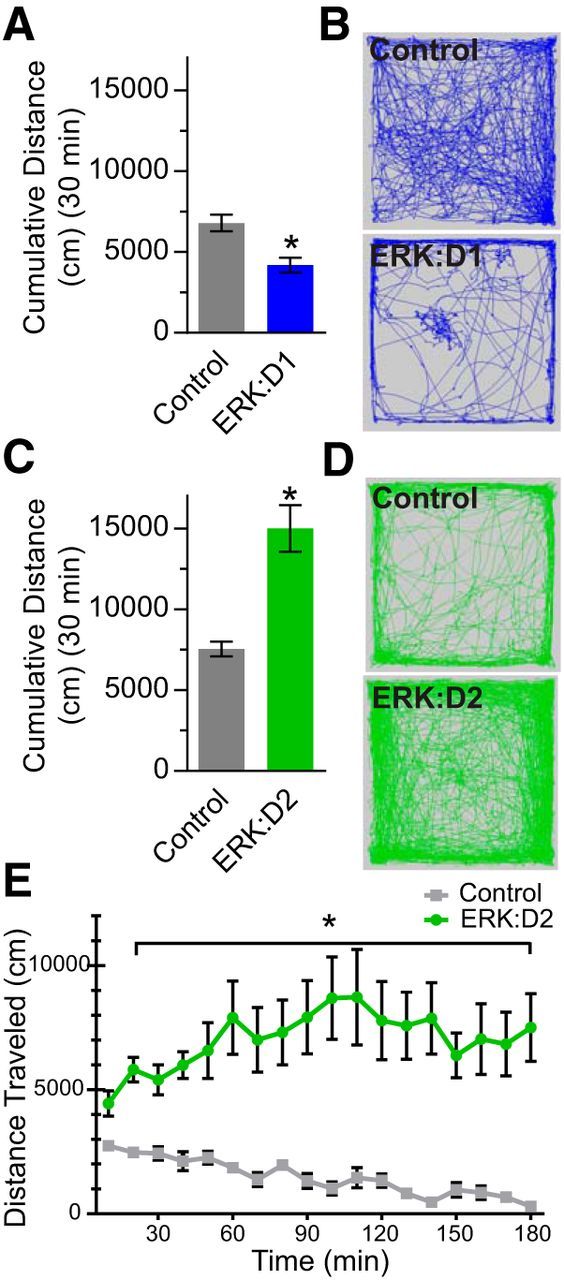
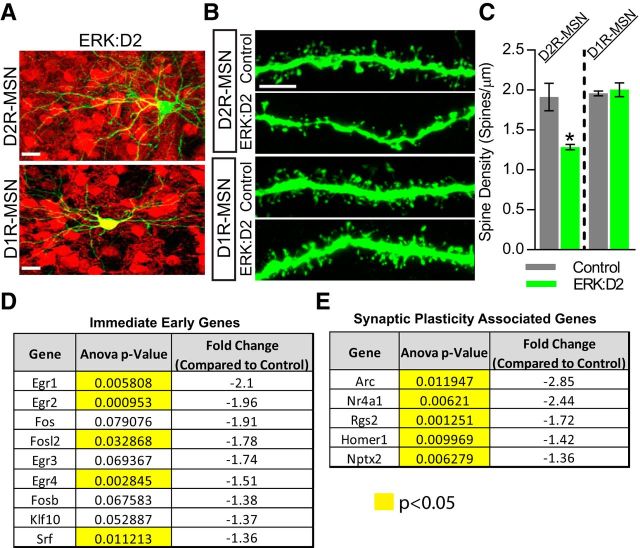
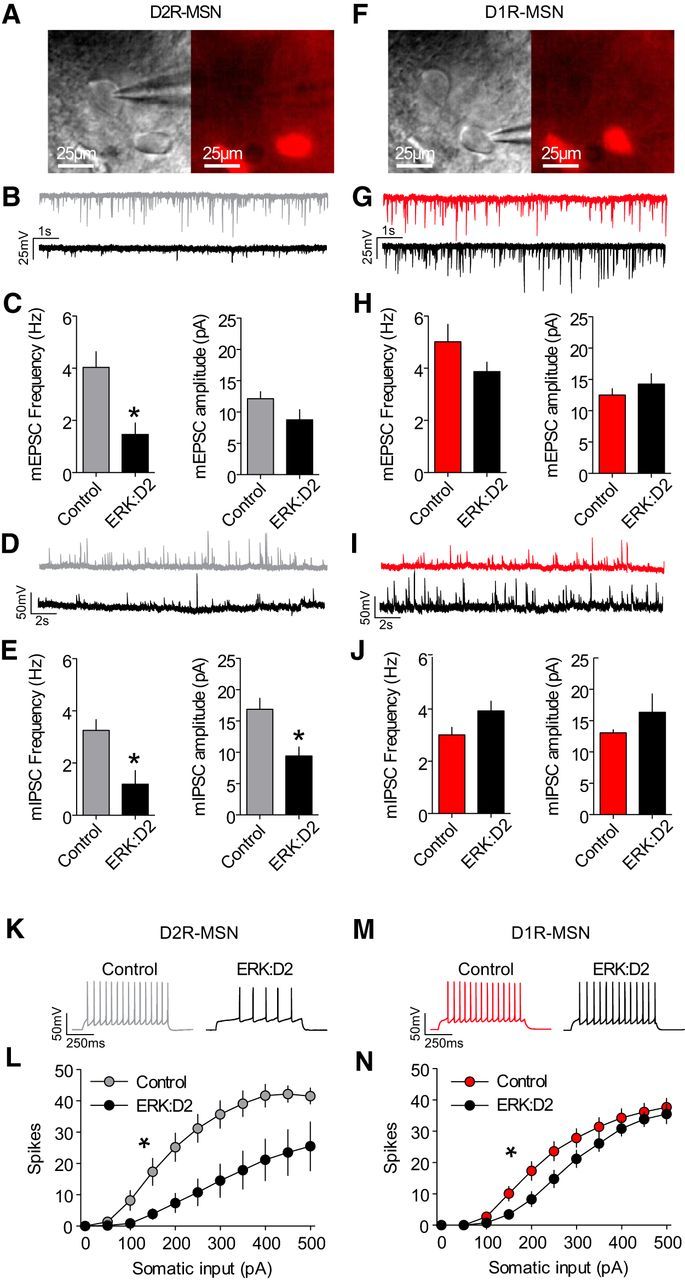
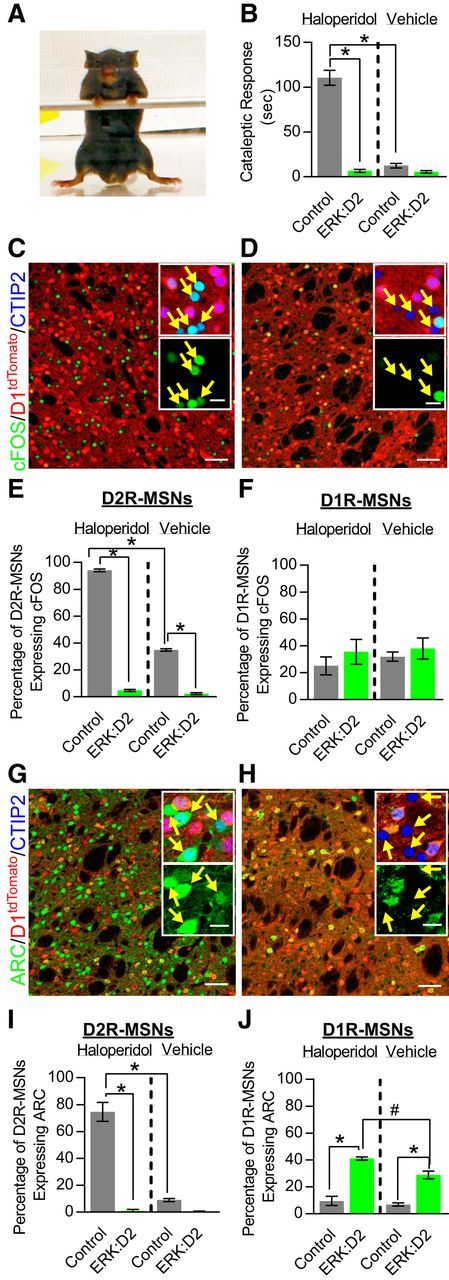
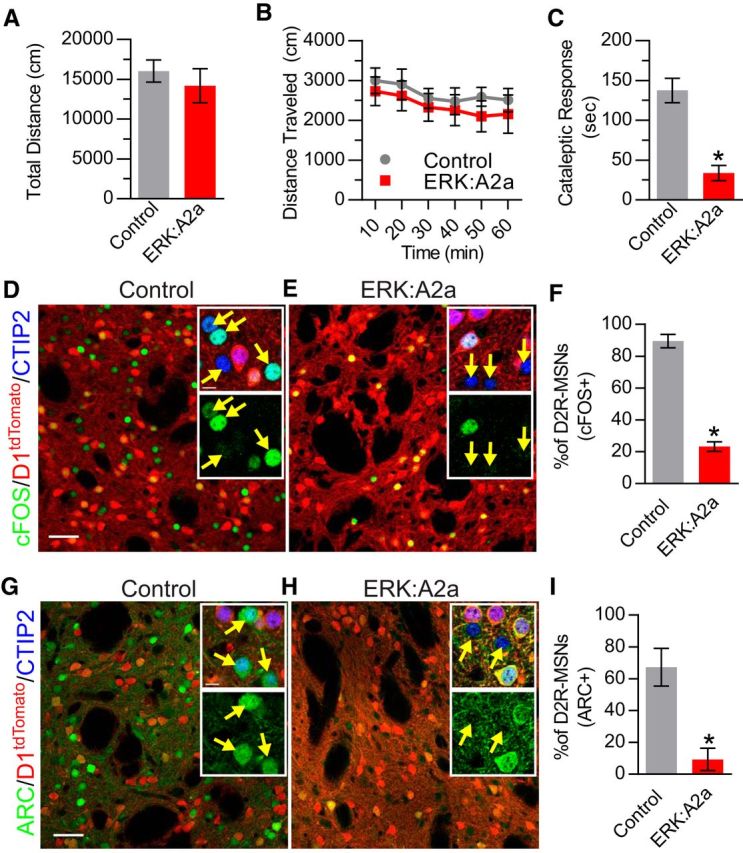
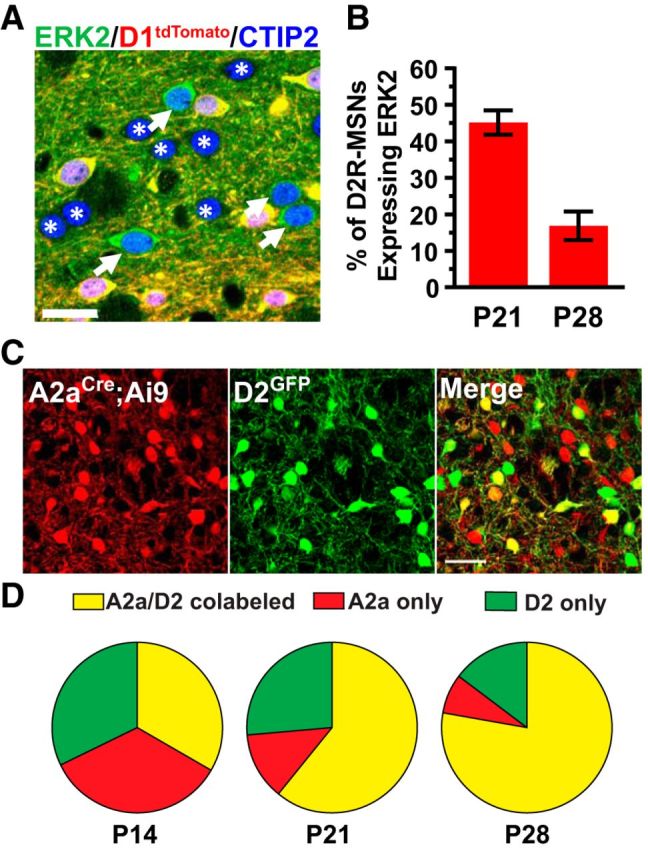
The ERK/MAPK intracellular signaling pathway is hypothesized to be a key regulator of striatal activity via modulation of synaptic plasticity and gene transcription. However, prior investigations into striatal ERK/MAPK functions have yielded conflicting results. Further, these studies have not delineated the cell-type-specific roles of ERK/MAPK signaling due to the reliance on globally administered pharmacological ERK/MAPK inhibitors and the use of genetic models that only partially reduce total ERK/MAPK activity. Here, we generated mouse models in which ERK/MAPK signaling was completely abolished in each of the two distinct classes of medium spiny neurons (MSNs). ERK/MAPK deletion in D1R-MSNs (direct pathway) resulted in decreased locomotor behavior, reduced weight gain, and early postnatal lethality. In contrast, loss of ERK/MAPK signaling in D2R-MSNs (indirect pathway) resulted in a profound hyperlocomotor phenotype. ERK/MAPK-deficient D2R-MSNs exhibited a significant reduction in dendritic spine density, markedly suppressed electrical excitability, and suppression of activity-associated gene expression even after pharmacological stimulation. Our results demonstrate the importance of ERK/MAPK signaling in governing the motor functions of the striatal direct and indirect pathways. Our data further show a critical role for ERK in maintaining the excitability and plasticity of D2R-MSNs.SIGNIFICANCE STATEMENT Alterations in ERK/MAPK activity are associated with drug abuse, as well as neuropsychiatric and movement disorders. However, genetic evidence defining the functions of ERK/MAPK signaling in striatum-related neurophysiology and behavior is lacking. We show that loss of ERK/MAPK signaling leads to pathway-specific alterations in motor function, reduced neuronal excitability, and the inability of medium spiny neurons to regulate activity-induced gene expression. Our results underscore the potential importance of the ERK/MAPK pathway in human movement disorders.
Keywords: MAP kinase; activity-induced gene expression; basal ganglia; dendritic spine; hyperlocomotion; medium spiny neuron.
Copyright © 2017 the authors 0270-6474/17/378102-14$15.00/0.
Figures
References
-
- Adams JP, Anderson AE, Varga AW, Dineley KT, Cook RG, Pfaffinger PJ, Sweatt JD (2000) The A-type potassium channel Kv4.2 is a substrate for the mitogen-activated protein kinase ERK. J Neurochem 75:2277–2287. - PubMed
-
- Bertran-Gonzalez J, Bosch C, Maroteaux M, Matamales M, Hervé D, Valjent E, Girault JA (2008) Opposing patterns of signaling activation in dopamine D1 and D2 receptor-expressing striatal neurons in response to cocaine and haloperidol. J Neurosci 28:5671–5685. 10.1523/JNEUROSCI.1039-08.2008 - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous