

Osteosarcoma: Molecular Pathogenesis and iPSC Modeling
- PMID: 28735817
- PMCID: PMC5558609
- DOI: 10.1016/j.molmed.2017.06.004
Osteosarcoma: Molecular Pathogenesis and iPSC Modeling
Abstract
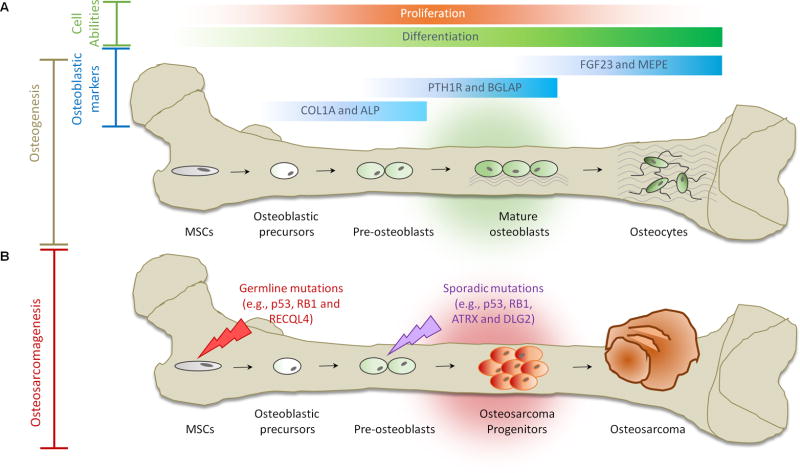
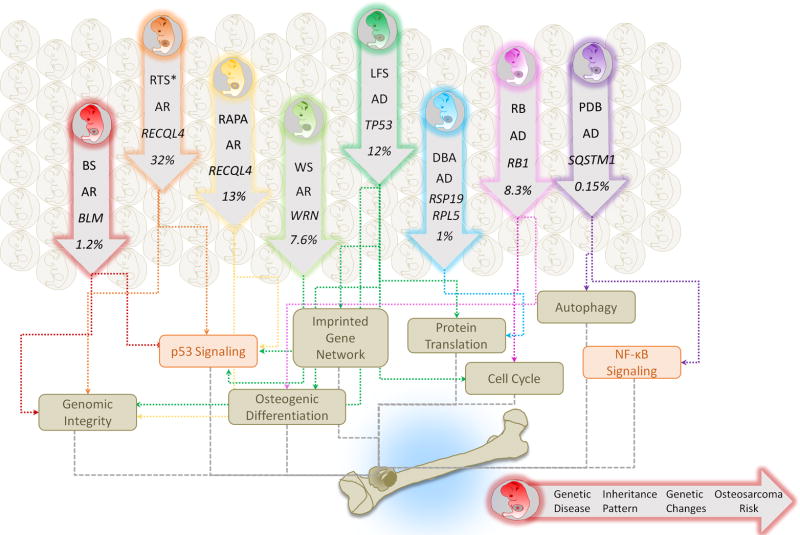
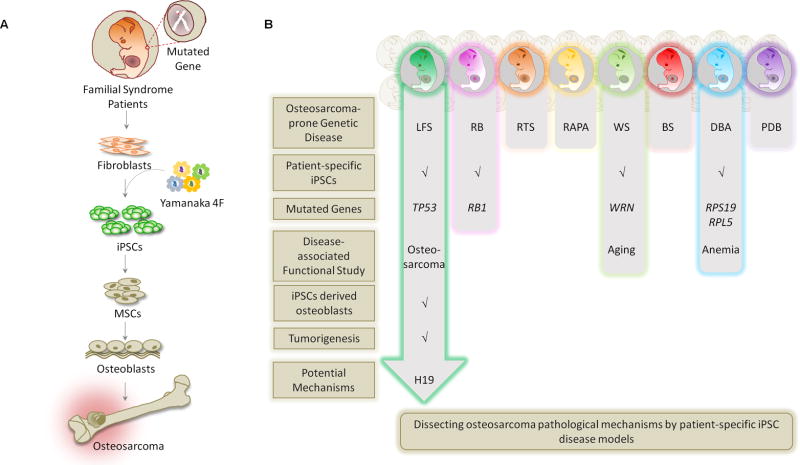
Rare hereditary disorders provide unequivocal evidence of the importance of genes in human disease pathogenesis. Familial syndromes that predispose to osteosarcomagenesis are invaluable in understanding the underlying genetics of this malignancy. Recently, patient-derived induced pluripotent stem cells (iPSCs) have been successfully utilized to model Li-Fraumeni syndrome (LFS)-associated bone malignancy, demonstrating that iPSCs can serve as an in vitro disease model to elucidate osteosarcoma etiology. We provide here an overview of osteosarcoma predisposition syndromes and review recently established iPSC disease models for these familial syndromes. Merging molecular information gathered from these models with the current knowledge of osteosarcoma biology will help us to gain a deeper understanding of the pathological mechanisms underlying osteosarcomagenesis and will potentially aid in the development of future patient therapies.
Keywords: cancer etiology; familial cancer syndrome; induced pluripotent stem cell; osteosarcoma.
Copyright © 2017 Elsevier Ltd. All rights reserved.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
