

Astrocytic tight junctions control inflammatory CNS lesion pathogenesis
- PMID: 28737509
- PMCID: PMC5531407
- DOI: 10.1172/JCI91301
Astrocytic tight junctions control inflammatory CNS lesion pathogenesis
Abstract
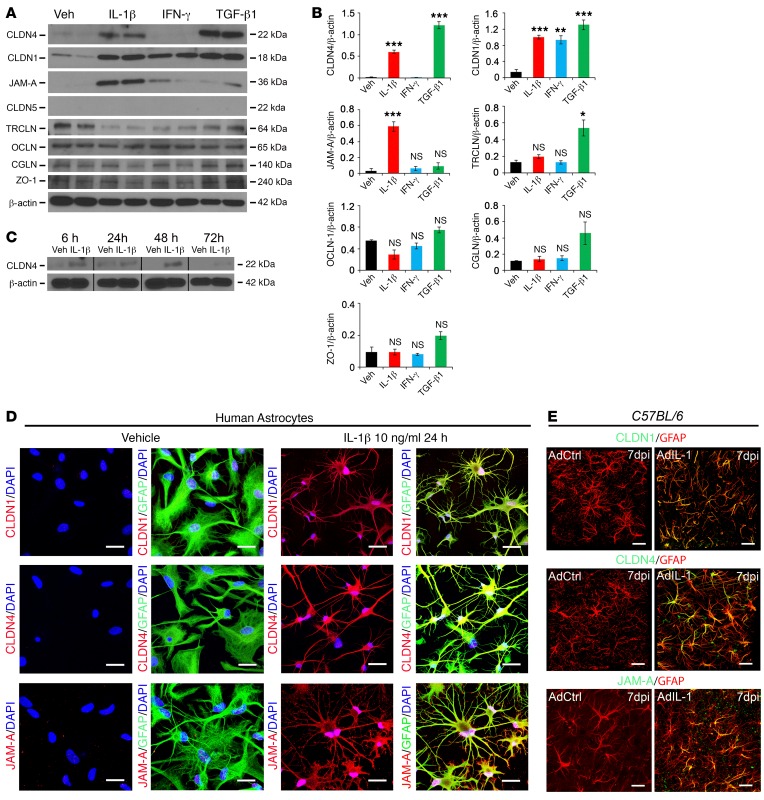
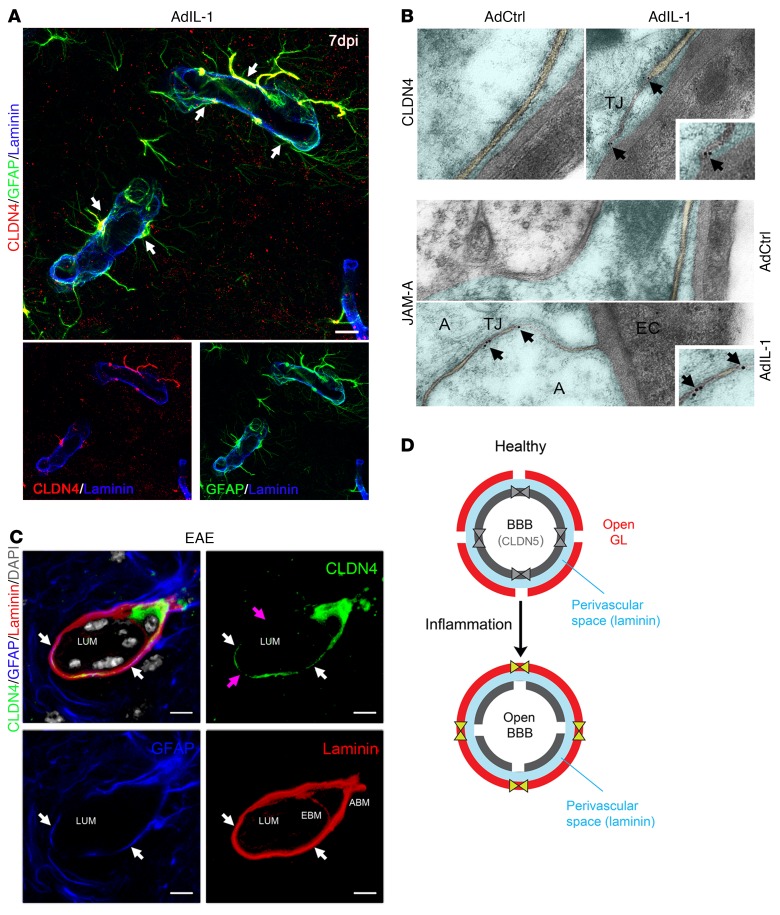
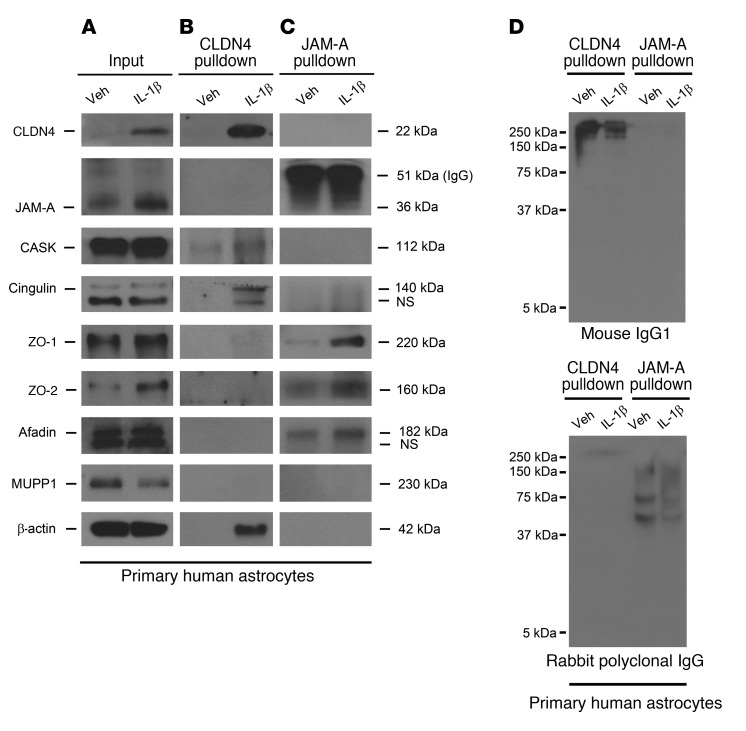
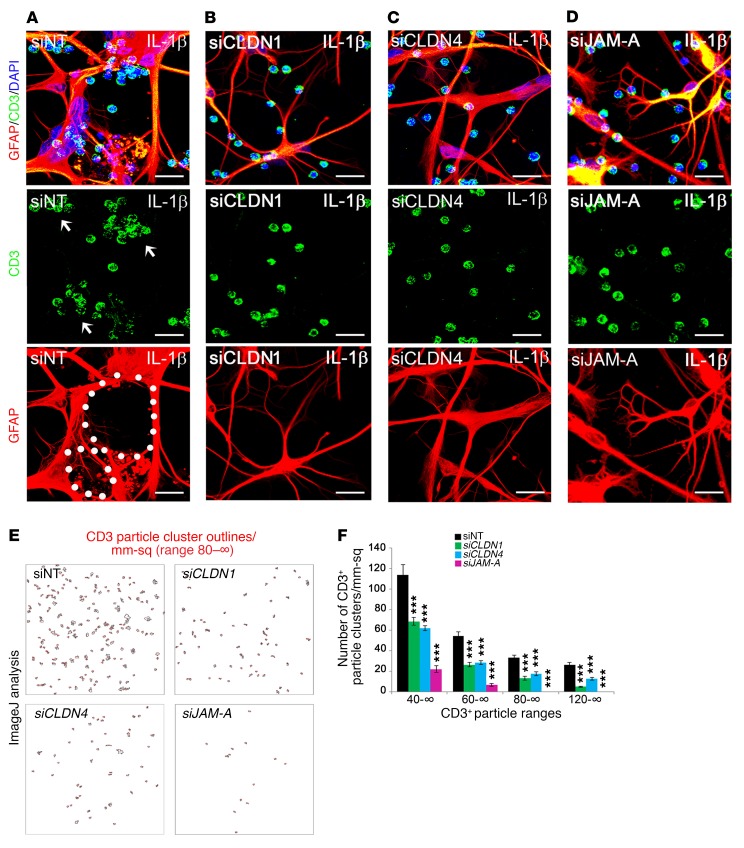
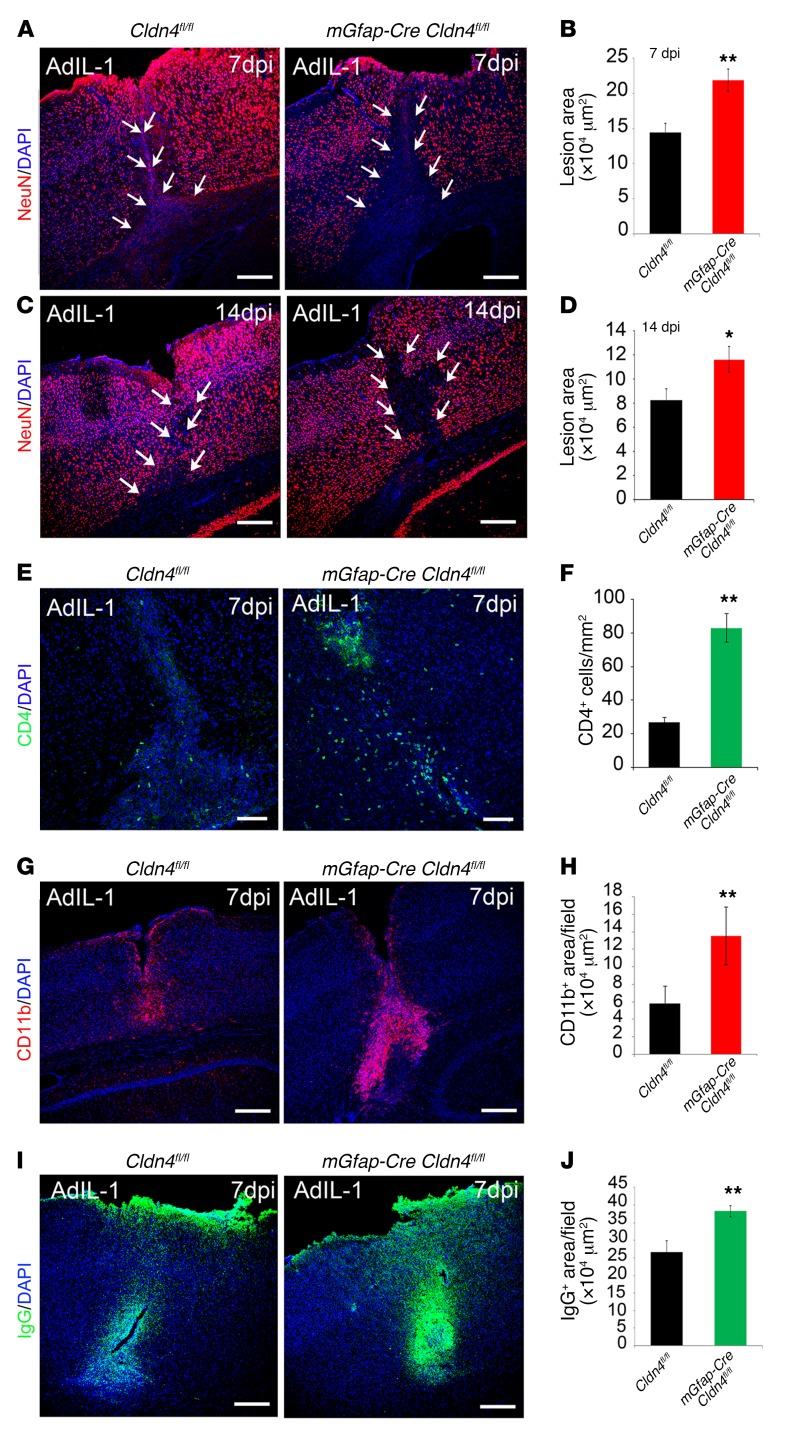
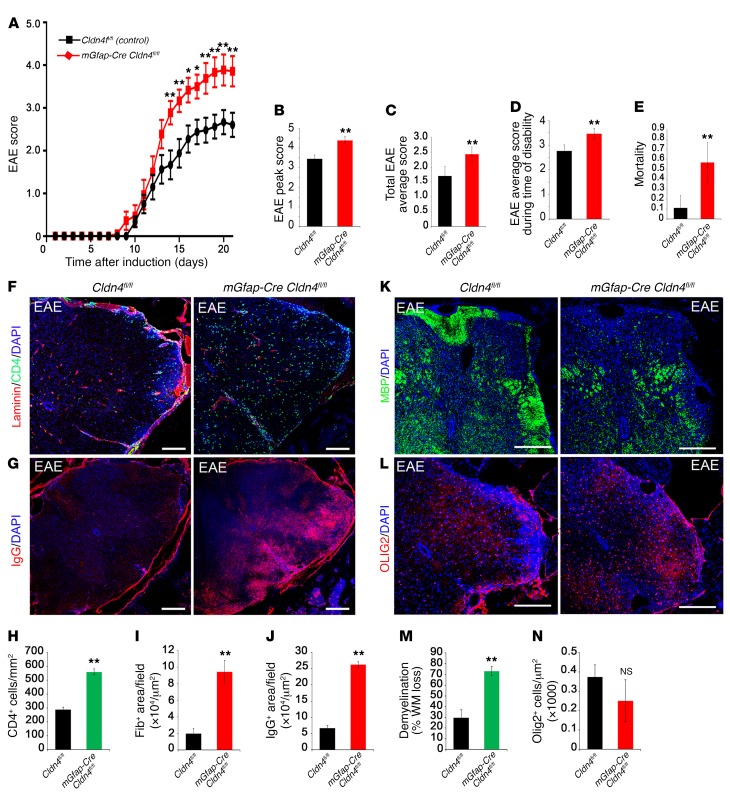
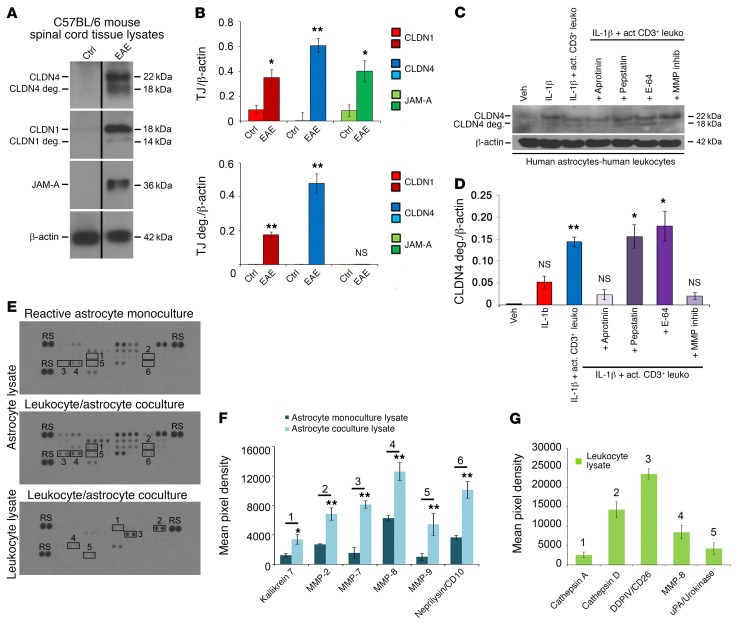
Lesions and neurologic disability in inflammatory CNS diseases such as multiple sclerosis (MS) result from the translocation of leukocytes and humoral factors from the vasculature, first across the endothelial blood-brain barrier (BBB) and then across the astrocytic glia limitans (GL). Factors secreted by reactive astrocytes open the BBB by disrupting endothelial tight junctions (TJs), but the mechanisms that control access across the GL are unknown. Here, we report that in inflammatory lesions, a second barrier composed of reactive astrocyte TJs of claudin 1 (CLDN1), CLDN4, and junctional adhesion molecule A (JAM-A) subunits is induced at the GL. In a human coculture model, CLDN4-deficient astrocytes were unable to control lymphocyte segregation. In models of CNS inflammation and MS, mice with astrocyte-specific Cldn4 deletion displayed exacerbated leukocyte and humoral infiltration, neuropathology, motor disability, and mortality. These findings identify a second inducible barrier to CNS entry at the GL. This barrier may be therapeutically targetable in inflammatory CNS disease.
Conflict of interest statement
Figures
Comment in
- Astrocytes to the rescue! Glia limitans astrocytic endfeet control CNS inflammation doi: 10.1172/JCI95769
References
-
- Miller DH, et al. Serial gadolinium enhanced magnetic resonance imaging in multiple sclerosis. Brain. 1988;111(Pt 4):927–939. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
