

Cardiac amyloidosis: An update on pathophysiology, diagnosis, and treatment
- PMID: 28739313
- PMCID: PMC5741539
- DOI: 10.1016/j.tcm.2017.07.004
Cardiac amyloidosis: An update on pathophysiology, diagnosis, and treatment
Abstract
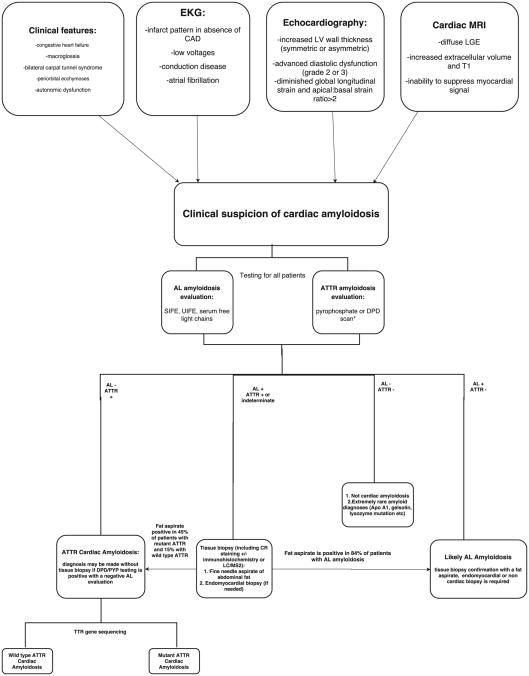
The amyloidoses are a group of systemic diseases characterized by organ deposition of misfolded protein fragments of diverse origins. The natural history of the disease, involvement of other organs, and treatment options vary significantly based on the protein of origin. In AL amyloidosis, amyloid protein is derived from immunoglobulin light chains, and most often involves the kidneys and the heart. ATTR amyloidosis is categorized as mutant or wild-type depending on the genetic sequence of the transthyretin (TTR) protein produced by the liver. Wild-type ATTR amyloidosis mainly involves the heart, although the reported occurrence of bilateral carpal tunnel syndrome, spinal stenosis and biceps tendon rupture in these patients speaks to more generalized protein deposition. Mutant TTR is marked by cardiac and/or peripheral nervous system involvement. Cardiac involvement is associated with symptoms of heart failure, and dictates the clinical course of the disease. Cardiac amyloidosis can be diagnosed noninvasively by echocardiography, cardiac MRI, or nuclear scintigraphy. Endomyocardial biopsy may be needed in the case of equivocal imaging findings or discordant data. Treatment is aimed at relieving congestive symptoms and targeting the underlying amyloidogenic process. This includes anti-plasma cell therapy in AL amyloidosis, and stabilization of the TTR tetramer or inhibition of TTR protein production in ATTR amyloidosis. Cardiac transplantation can be considered in highly selected patients in tandem with therapy aimed at suppressing the amyloidogenic process, and appears associated with durable long-term survival.
Keywords: AL amyloidosis; Restrictive cardiomyopathy; Transthyretin amyloidosis.
Copyright © 2018 Elsevier Inc. All rights reserved.
Figures
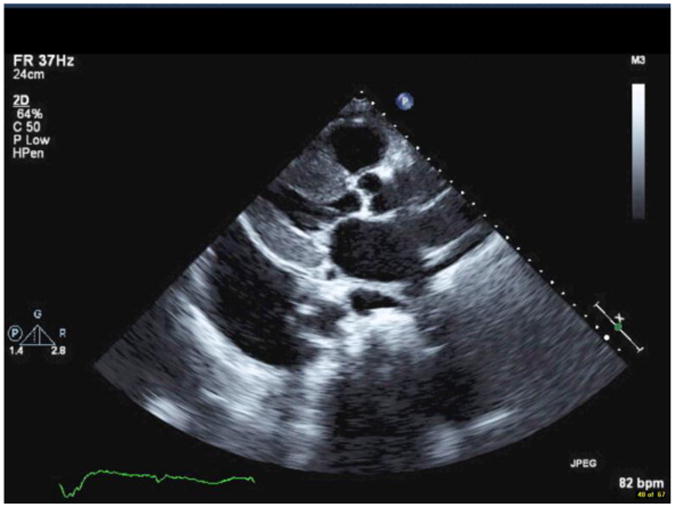
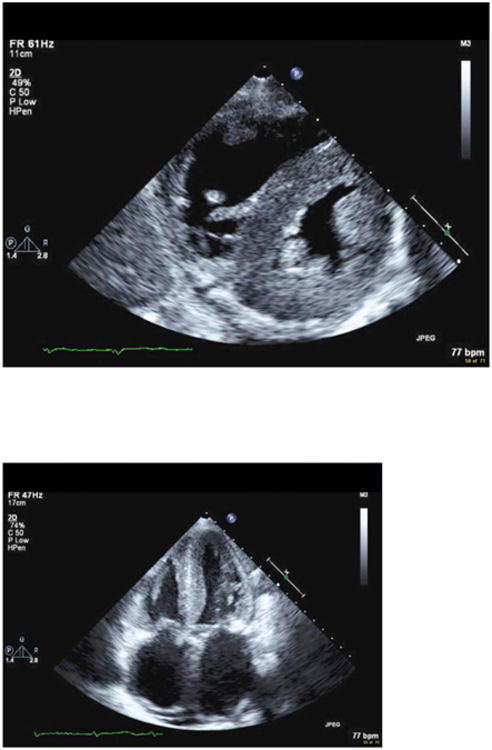
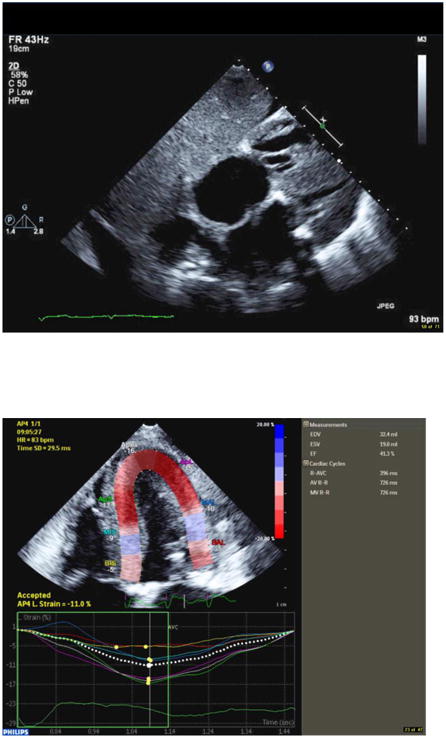
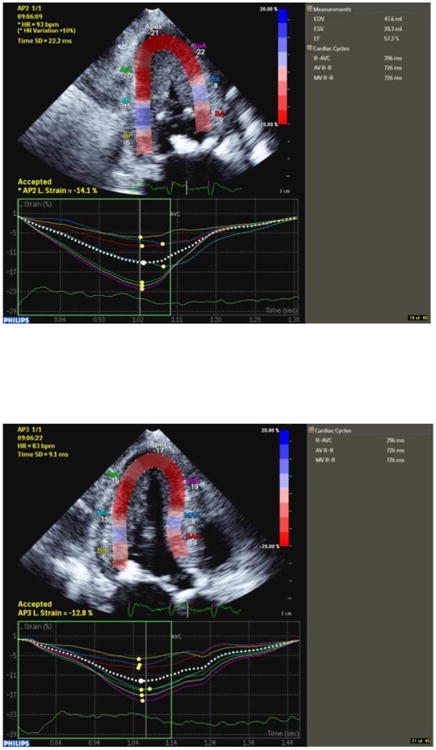
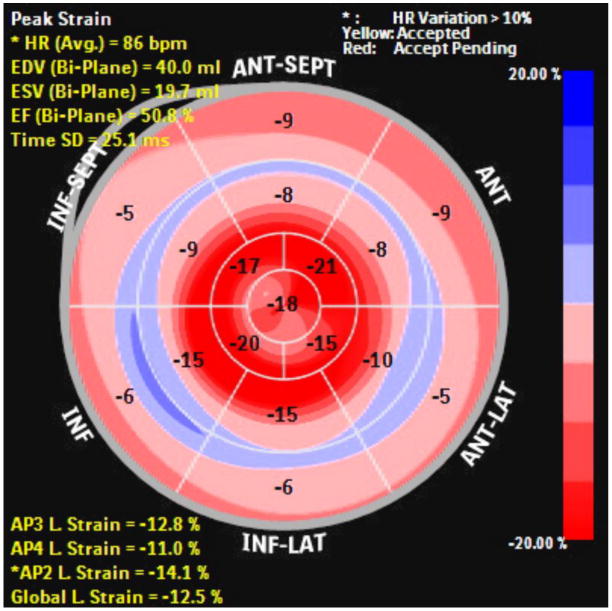
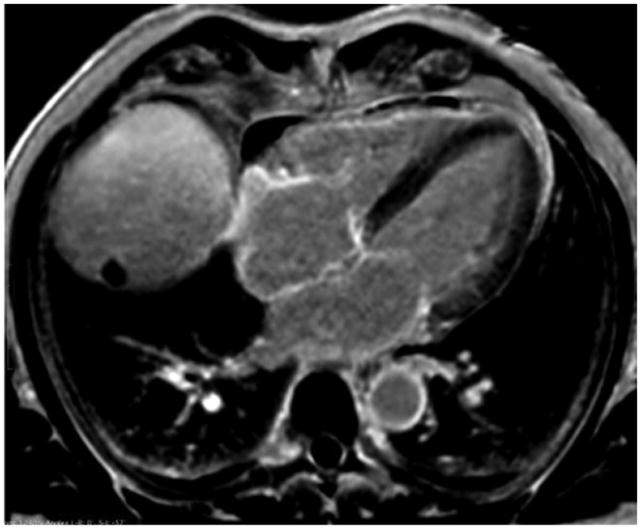
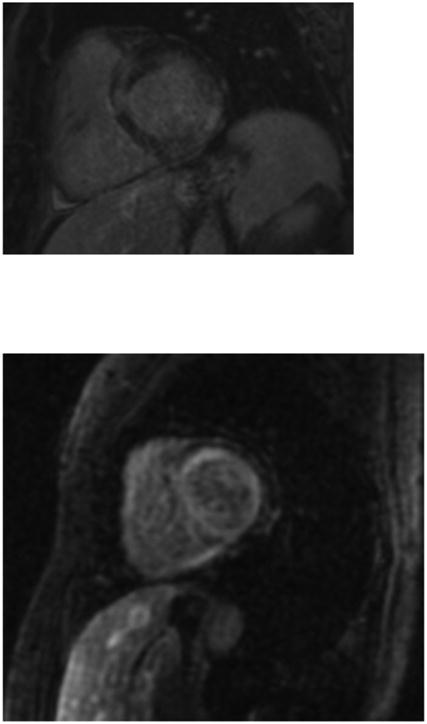

Comment in
-
Editorial commentary: Cardiac amyloidosis-Reversing the mindset and the cardiomyopathy.Trends Cardiovasc Med. 2018 Jan;28(1):22-23. doi: 10.1016/j.tcm.2017.07.005. Epub 2017 Jul 18. Trends Cardiovasc Med. 2018. PMID: 28754231 No abstract available.
References
-
- Sipe JD, Cohen AS. Review: history of the amyloid fibril. J Struct Biol. 2000;130(2-3):88–98. - PubMed
-
- Falk RH, Alexander KM, Liao R, Dorbala S. AL (Light-Chain) Cardiac Amyloidosis: A Review of Diagnosis and Therapy. Journal of the American College of Cardiology. 2016;68(12):1323–41. - PubMed
-
- Gertz MA, Comenzo R, Falk RH, Fermand JP, Hazenberg BP, Hawkins PN, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18-22 April 2004. American journal of hematology. 2005;79(4):319–28. - PubMed
-
- Skinner M, Cohen AS. The prealbumin nature of the amyloid protein in familial amyloid polyneuropathy (FAP)-swedish variety. Biochemical and biophysical research communications. 1981;99(4):1326–32. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
