

Fragile sites in cancer: more than meets the eye
- PMID: 28740117
- PMCID: PMC5546318
- DOI: 10.1038/nrc.2017.52
Fragile sites in cancer: more than meets the eye
Abstract
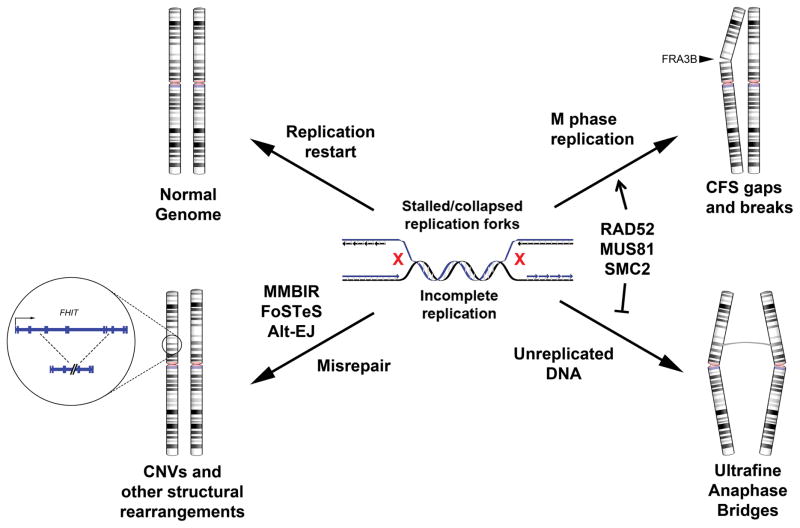
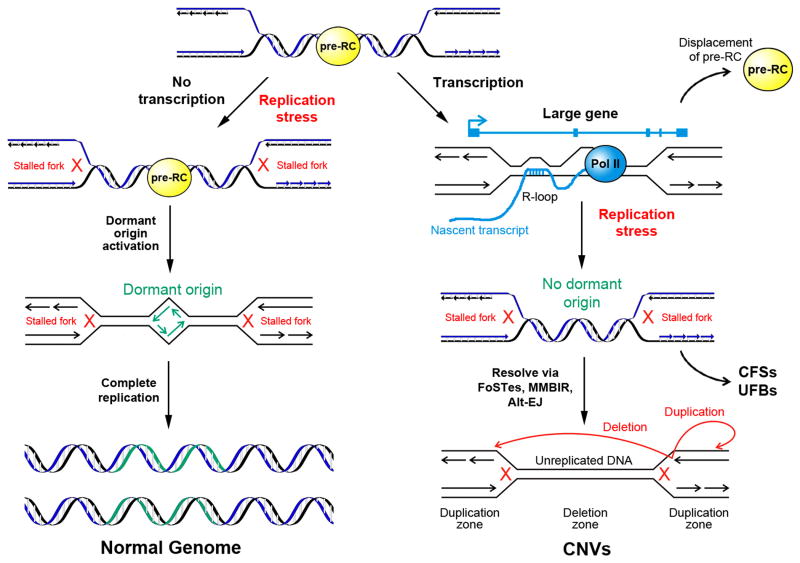
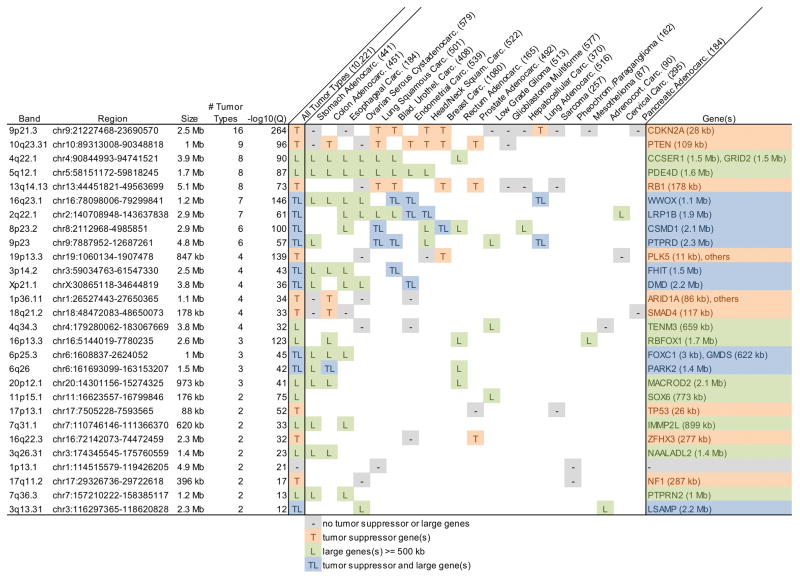
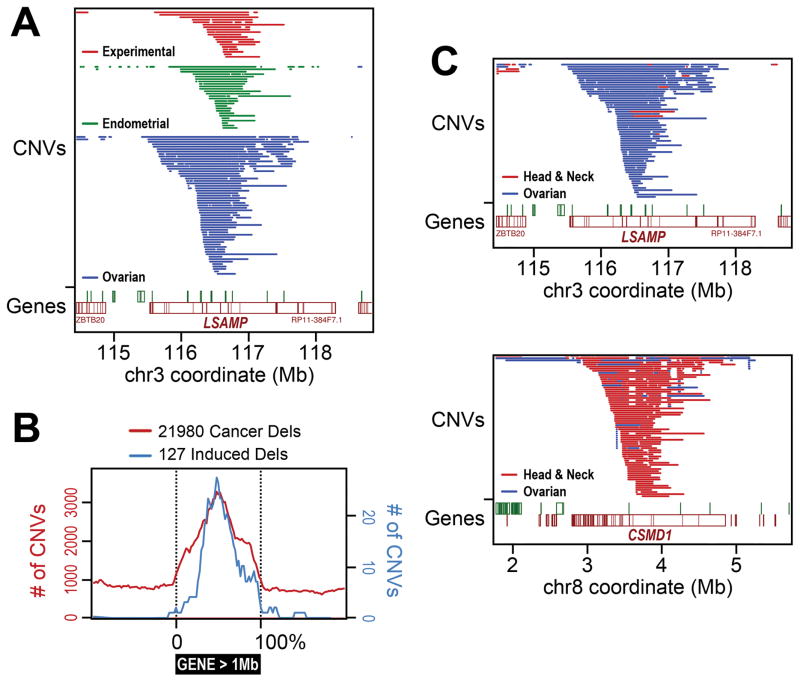
Ever since initial suggestions that instability at common fragile sites (CFSs) could be responsible for chromosome rearrangements in cancers, CFSs and associated genes have been the subject of numerous studies, leading to questions and controversies about their role and importance in cancer. It is now clear that CFSs are not frequently involved in translocations or other cancer-associated recurrent gross chromosome rearrangements. However, recent studies have provided new insights into the mechanisms of CFS instability, their effect on genome instability, and their role in generating focal copy number alterations that affect the genomic landscape of many cancers.
Conflict of interest statement
There is
Figures
References
-
- Glover TW, Berger C, Coyle J, Echo B. DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum Genet. 1984;67:136–42. - PubMed
-
- Yunis JJ. Fragile sites and predisposition to leukemia and lymphoma. Cancer Genet Cytogenet. 1984;12:85–8. - PubMed
-
- de Braekeleer M. Fragile sites and chromosomal structural rearrangements in human leukemia and cancer. Anticancer Res. 1987;7:417–22. - PubMed
-
- Yunis JJ. Fragile sites, mutagens and genomic rearrangements in cancer. Basic Life Sci. 1988;43:11–21. - PubMed
-
- Le Beau MM. Chromosomal fragile sites and cancer-specific rearrangements. Blood. 1986;67:849–58. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
