

Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm
- PMID: 28740174
- PMCID: PMC5524637
- DOI: 10.1038/s41467-017-00127-0
Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm
Abstract
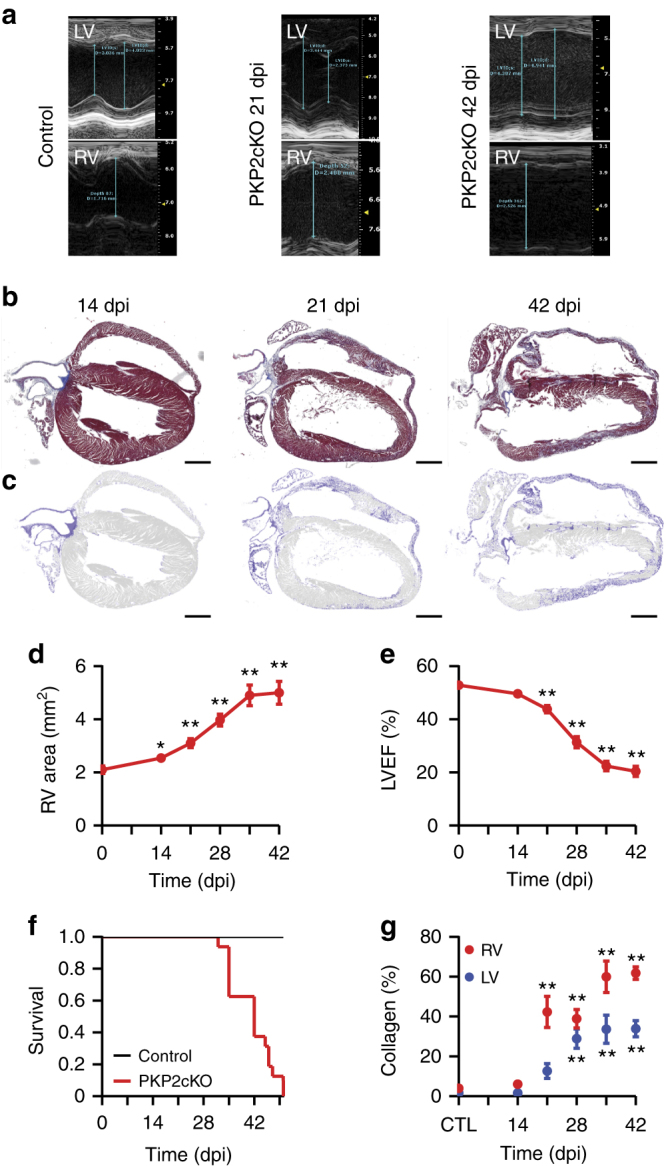
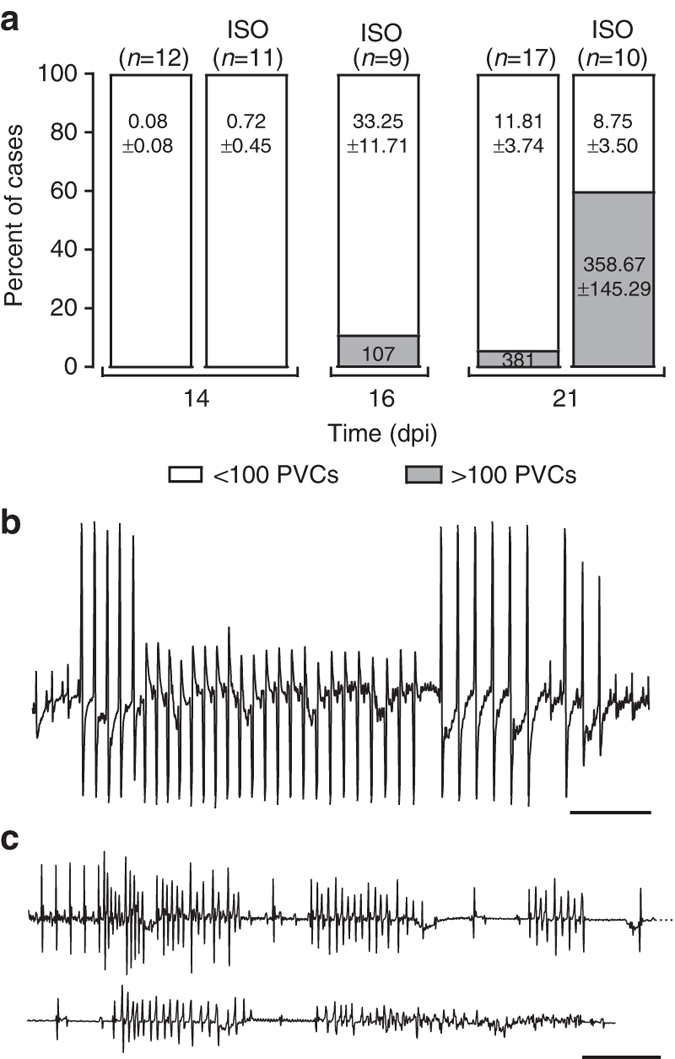
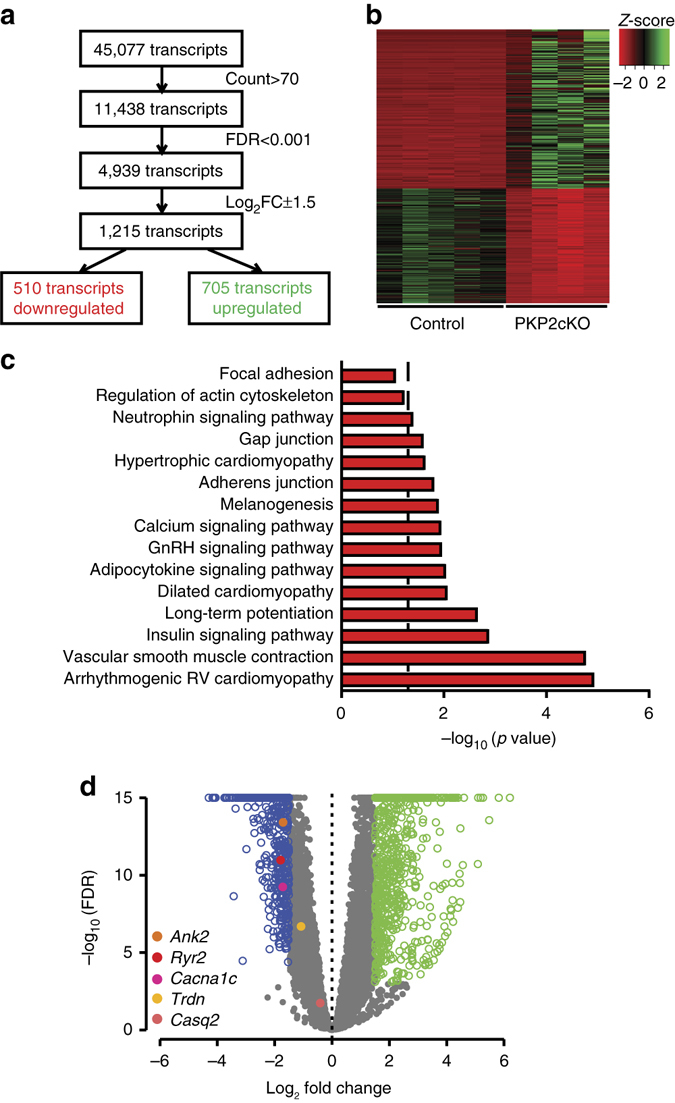
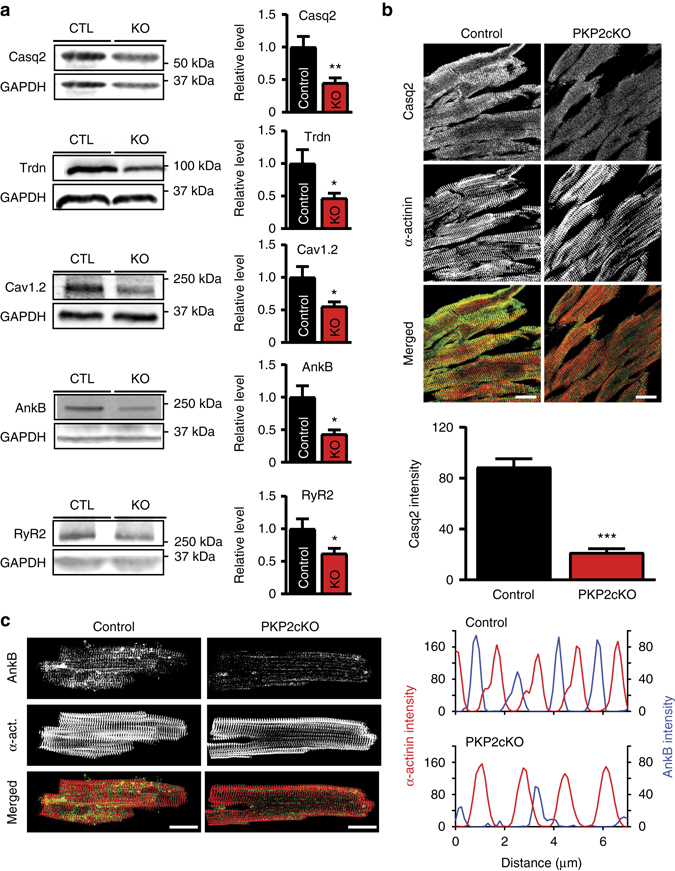
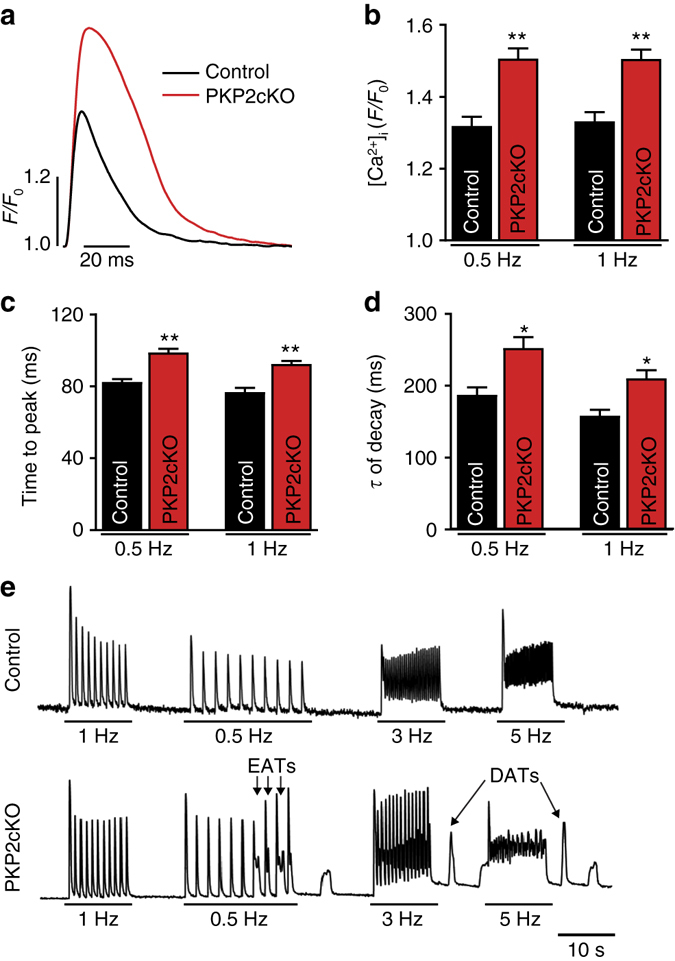
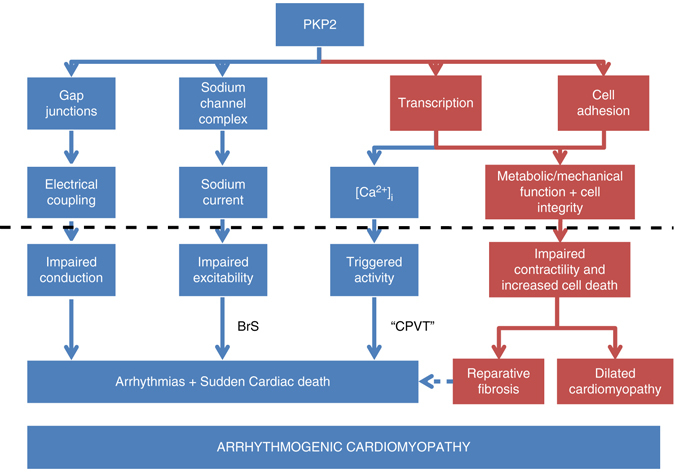
Plakophilin-2 (PKP2) is a component of the desmosome and known for its role in cell-cell adhesion. Mutations in human PKP2 associate with a life-threatening arrhythmogenic cardiomyopathy, often of right ventricular predominance. Here, we use a range of state-of-the-art methods and a cardiomyocyte-specific, tamoxifen-activated, PKP2 knockout mouse to demonstrate that in addition to its role in cell adhesion, PKP2 is necessary to maintain transcription of genes that control intracellular calcium cycling. Lack of PKP2 reduces expression of Ryr2 (coding for Ryanodine Receptor 2), Ank2 (coding for Ankyrin-B), Cacna1c (coding for CaV1.2) and Trdn (coding for triadin), and protein levels of calsequestrin-2 (Casq2). These factors combined lead to disruption of intracellular calcium homeostasis and isoproterenol-induced arrhythmias that are prevented by flecainide treatment. We propose a previously unrecognized arrhythmogenic mechanism related to PKP2 expression and suggest that mutations in PKP2 in humans may cause life-threatening arrhythmias even in the absence of structural disease.It is believed that mutations in desmosomal adhesion complex protein plakophilin 2 (PKP2) cause arrhythmia due to loss of cell-cell communication. Here the authors show that PKP2 controls the expression of proteins involved in calcium cycling in adult mouse hearts, and that lack of PKP2 can cause arrhythmia in a structurally normal heart.
Conflict of interest statement
M.J.A. is a consultant for Boston Scientific, Gilead Sciences, Invitae, Medtronic, MyoKardia, and St. Jude Medical. M.J.A., D.T., and Mayo Clinic have received sales-based royalties from Transgenomic for their FAMILION-LQTS and FAMILION-CPVT genetic tests. M.J.A. and Mayo Clinic have licensed intellectual property to AliveCor. The remaining authors declare no competing financial interests.
Figures
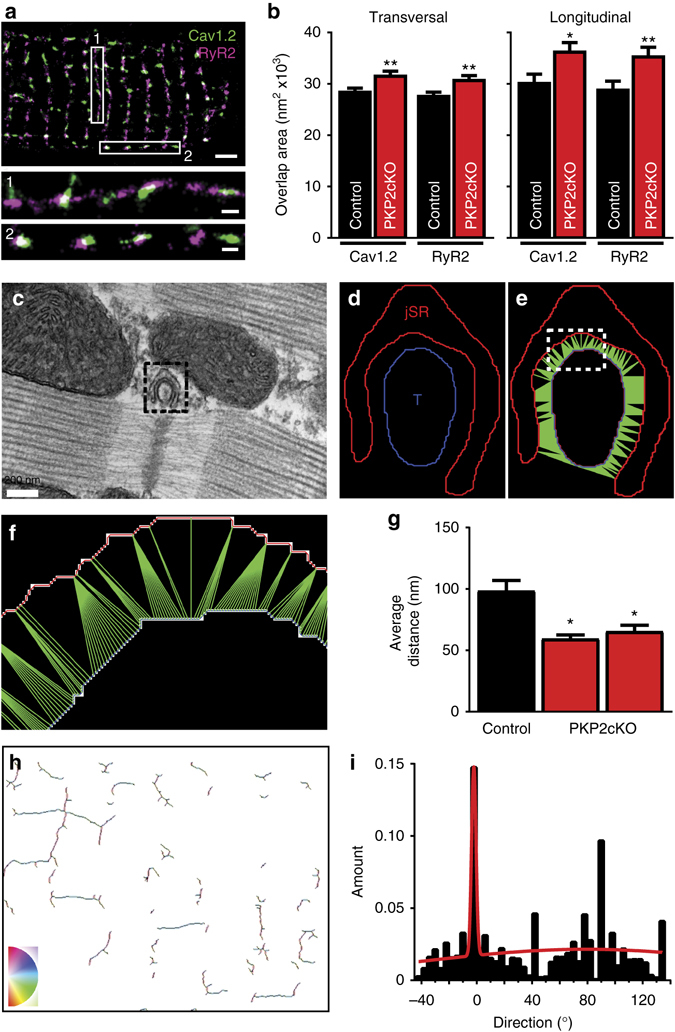
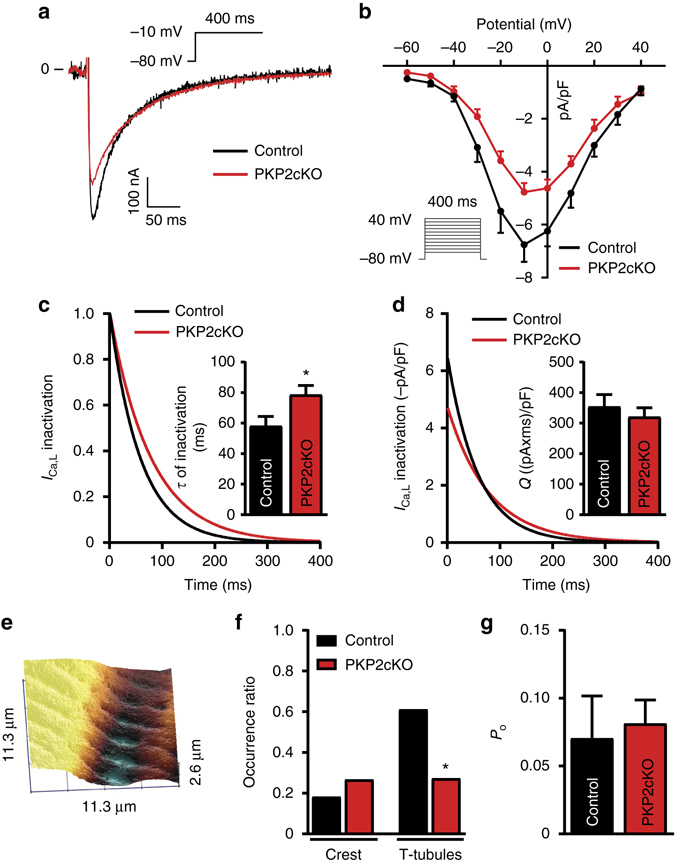
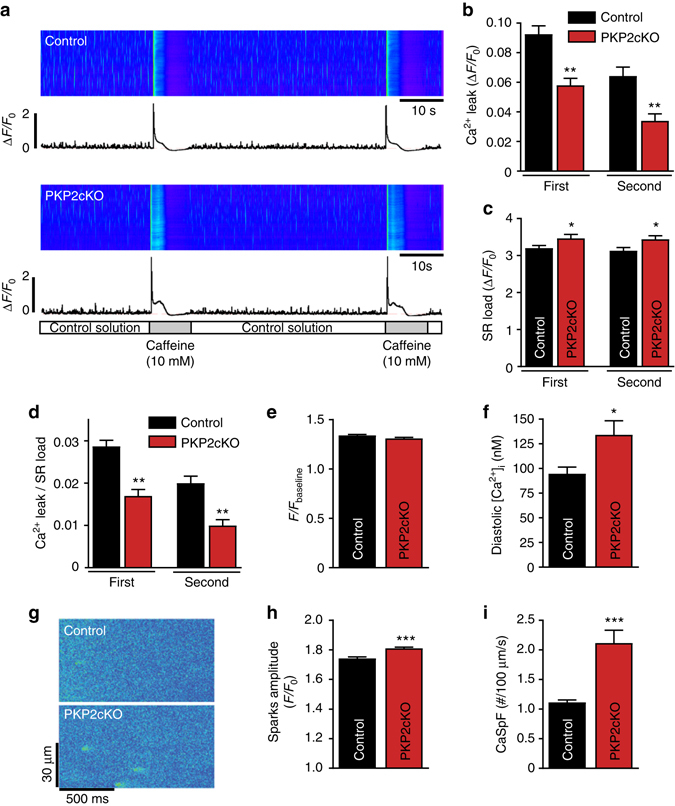
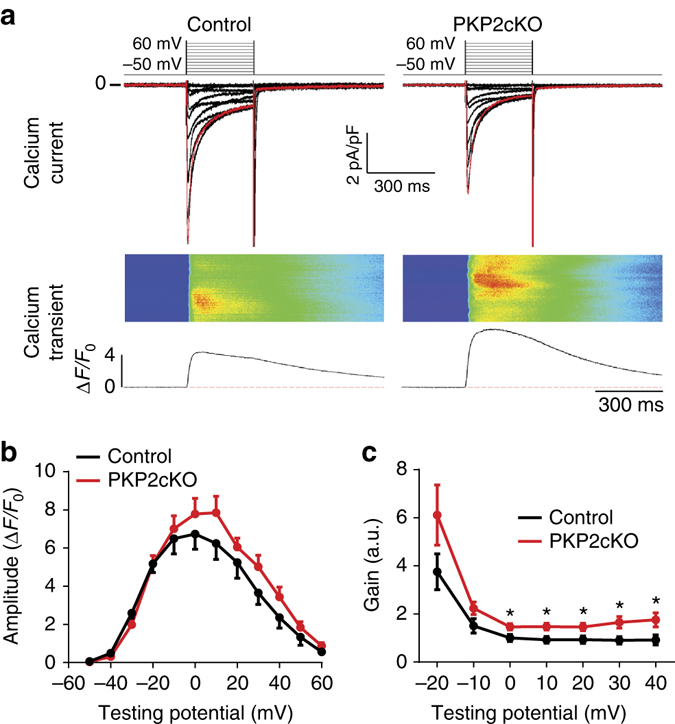
References
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL134344/HL/NHLBI NIH HHS/United States
- R21 CA187612/CA/NCI NIH HHS/United States
- R01 GM057691/GM/NIGMS NIH HHS/United States
- R01 HL076751/HL/NHLBI NIH HHS/United States
- R01 HL134328/HL/NHLBI NIH HHS/United States
- DP1 HL123271/HL/NHLBI NIH HHS/United States
- R01 HL055438/HL/NHLBI NIH HHS/United States
- R01 GM108119/GM/NIGMS NIH HHS/United States
- R01 HL126905/HL/NHLBI NIH HHS/United States
- R01 HL120108/HL/NHLBI NIH HHS/United States
- R01 HL126802/HL/NHLBI NIH HHS/United States
- P30 CA016087/CA/NCI NIH HHS/United States
- R01 HL136179/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
