

p62/SQSTM1/Sequestosome-1 is an N-recognin of the N-end rule pathway which modulates autophagosome biogenesis
- PMID: 28740232
- PMCID: PMC5524641
- DOI: 10.1038/s41467-017-00085-7
p62/SQSTM1/Sequestosome-1 is an N-recognin of the N-end rule pathway which modulates autophagosome biogenesis
Abstract
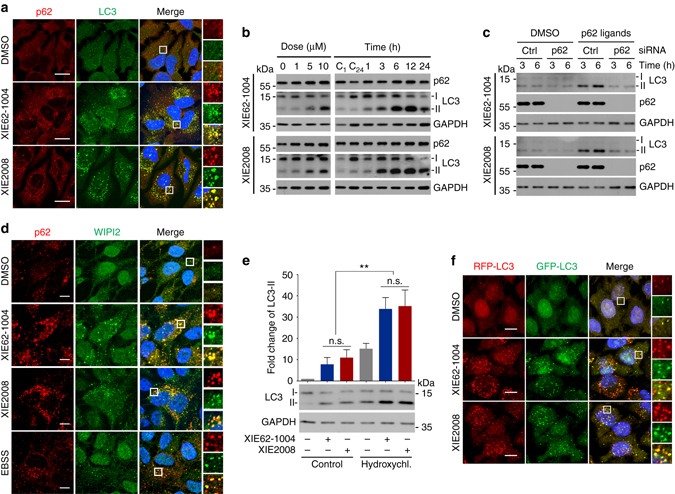
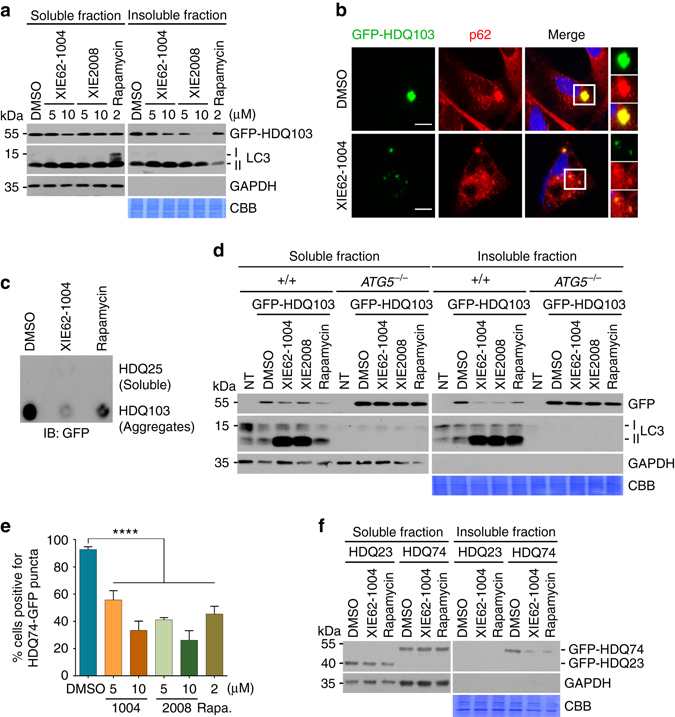
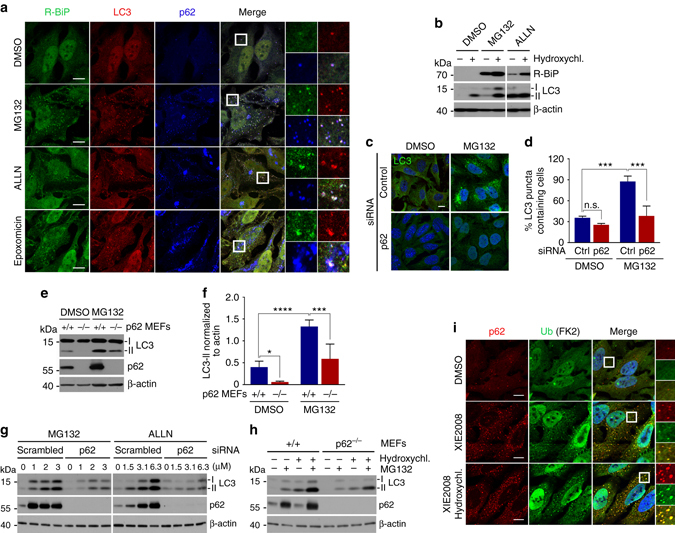
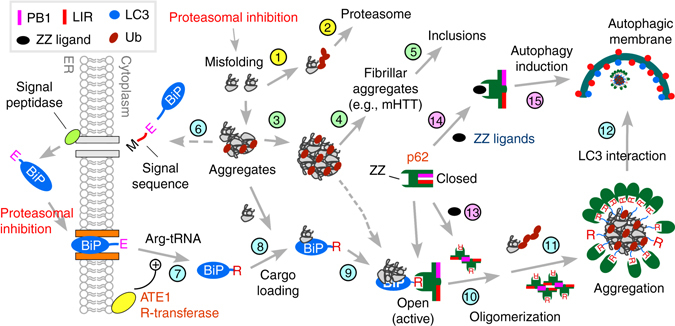
Macroautophagy mediates the selective degradation of proteins and non-proteinaceous cellular constituents. Here, we show that the N-end rule pathway modulates macroautophagy. In this mechanism, the autophagic adapter p62/SQSTM1/Sequestosome-1 is an N-recognin that binds type-1 and type-2 N-terminal degrons (N-degrons), including arginine (Nt-Arg). Both types of N-degrons bind its ZZ domain. By employing three-dimensional modeling, we developed synthetic ligands to p62 ZZ domain. The binding of Nt-Arg and synthetic ligands to ZZ domain facilitates disulfide bond-linked aggregation of p62 and p62 interaction with LC3, leading to the delivery of p62 and its cargoes to the autophagosome. Upon binding to its ligand, p62 acts as a modulator of macroautophagy, inducing autophagosome biogenesis. Through these dual functions, cells can activate p62 and induce selective autophagy upon the accumulation of autophagic cargoes. We also propose that p62 mediates the crosstalk between the ubiquitin-proteasome system and autophagy through its binding Nt-Arg and other N-degrons.Soluble misfolded proteins that fail to be degraded by the ubiquitin proteasome system (UPS) are redirected to autophagy via specific adaptors, such as p62. Here the authors show that p62 recognises N-degrons in these proteins, acting as a N-recognin from the proteolytic N-end rule pathway, and targets these cargos to autophagosomal degradation.
Conflict of interest statement
X.-Q.X. is a consultant for Oxis Biotech. The remaining authors declare no competing financial interests.
Figures
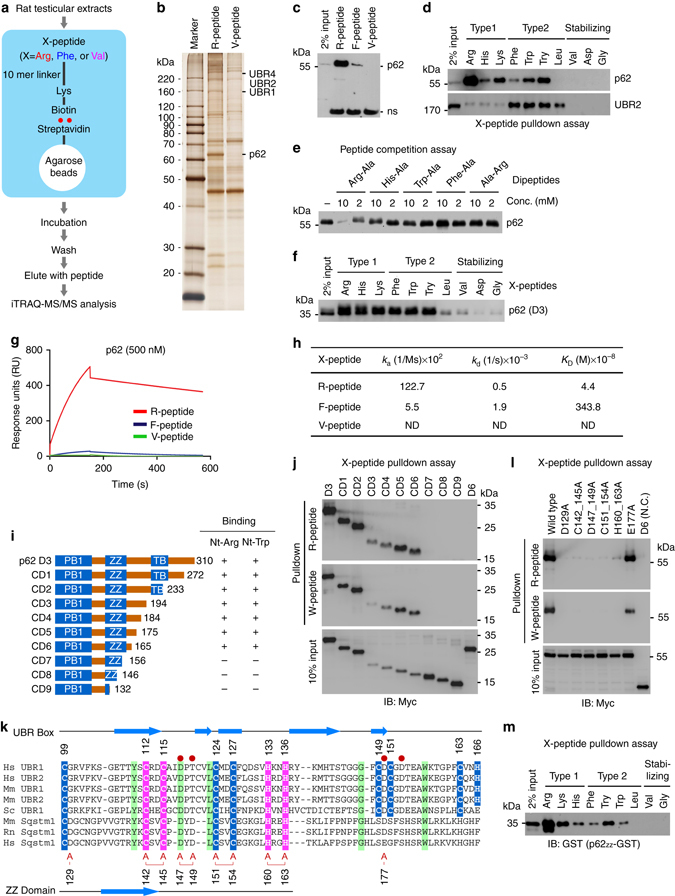
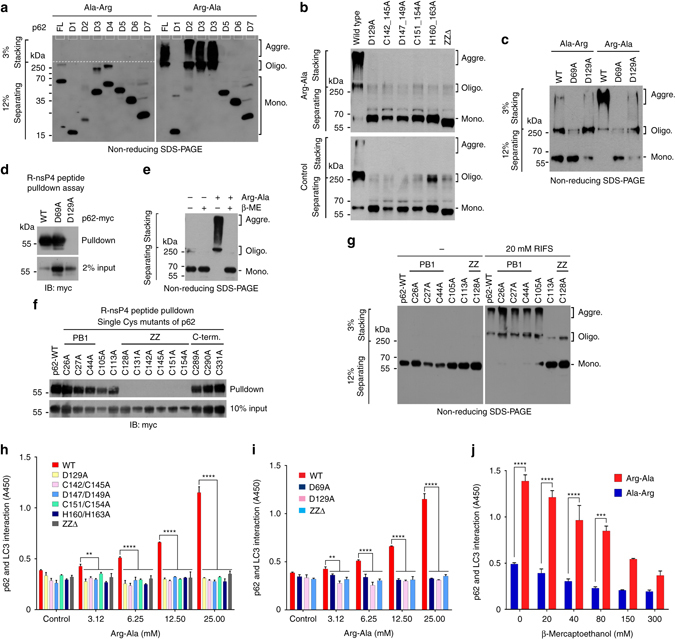
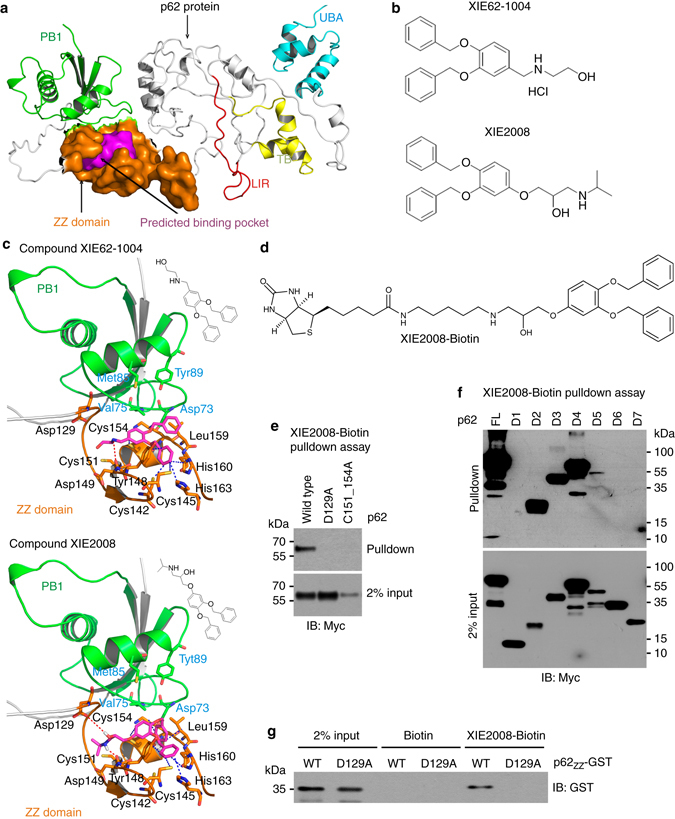
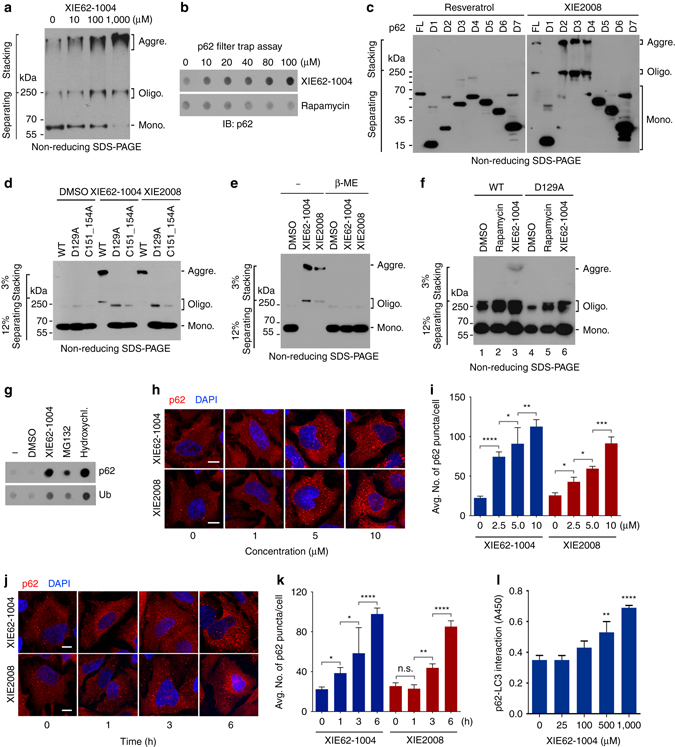
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
