

The 'Pushmi-Pullyu' of DNA REPAIR: Clinical Synthetic Lethality
- PMID: 28741503
- PMCID: PMC5527674
- DOI: 10.1016/j.trecan.2016.10.014
The 'Pushmi-Pullyu' of DNA REPAIR: Clinical Synthetic Lethality
Abstract
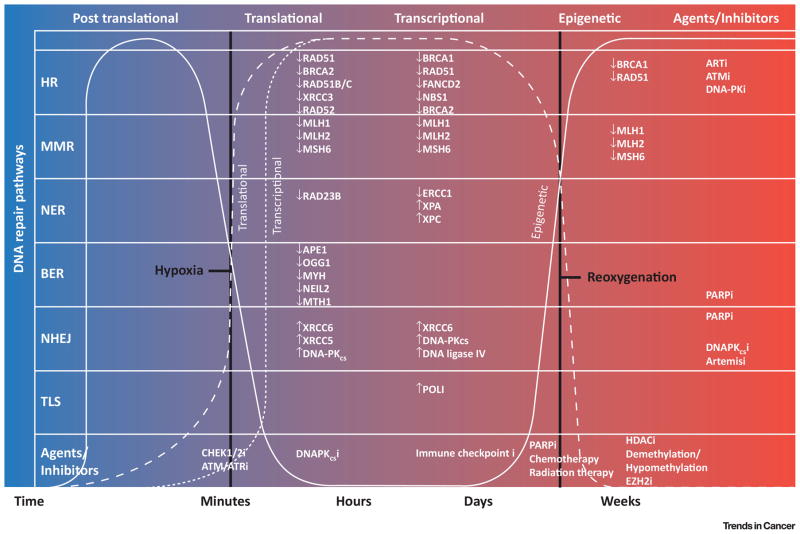
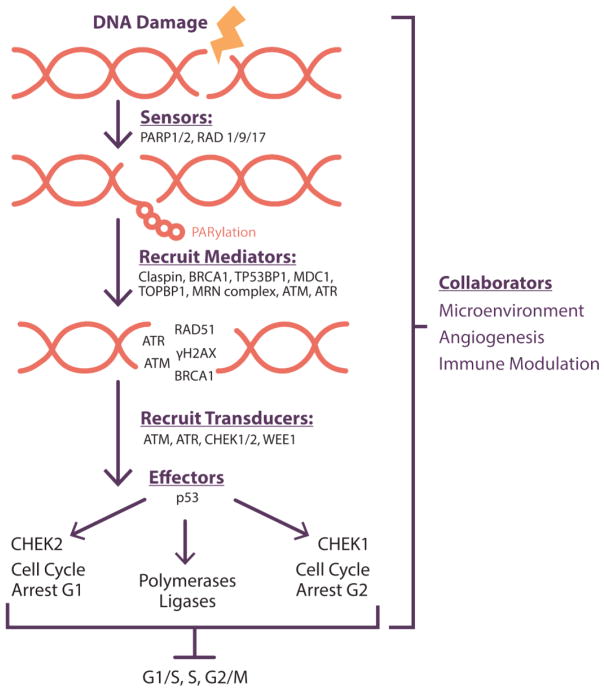
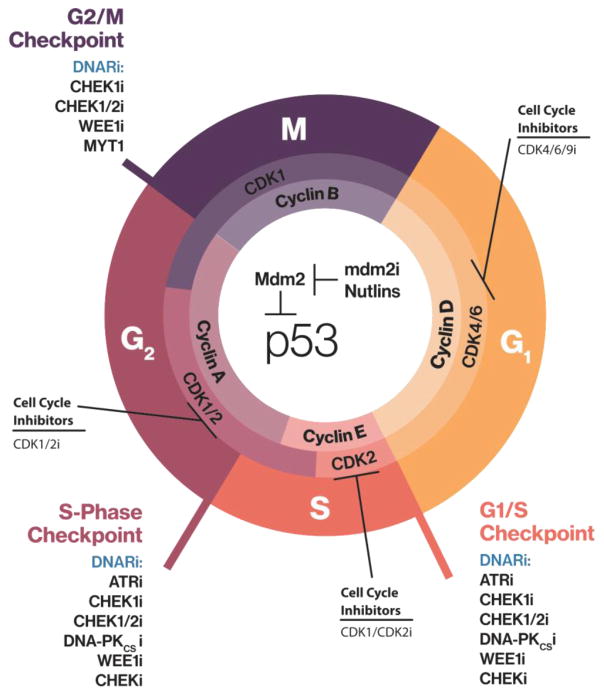
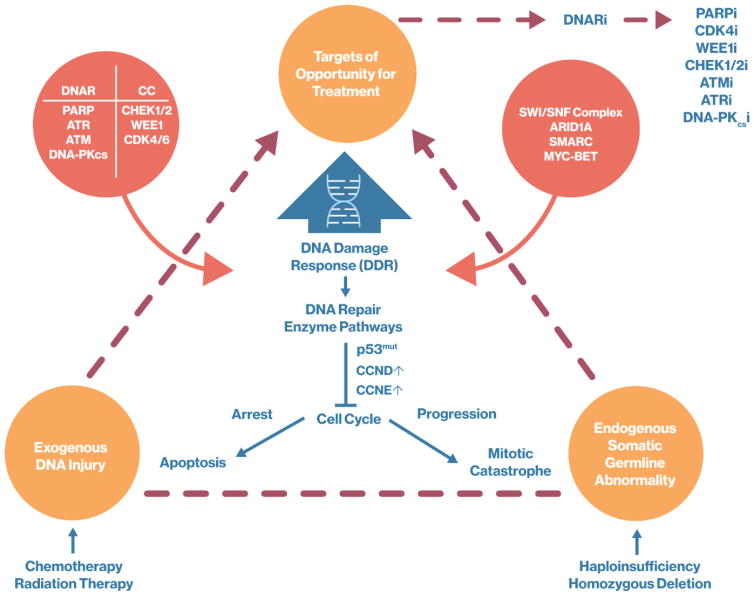
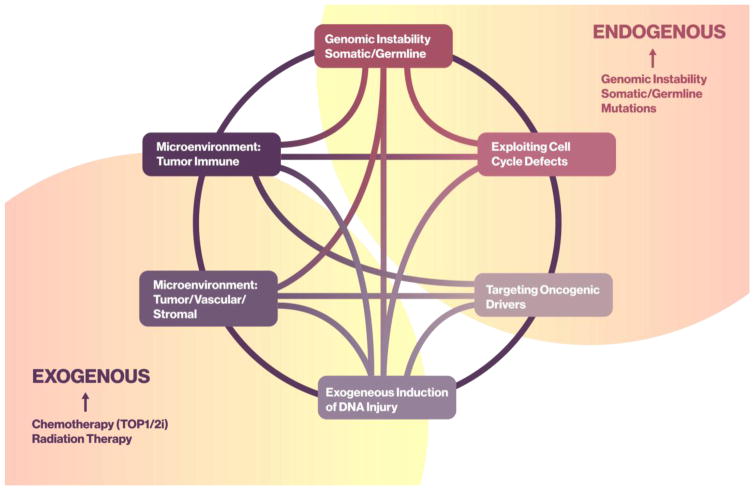
Maintenance of genomic integrity is critical for adaptive survival in the face of endogenous and exogenous environmental stress. The loss of stability and fidelity in the genome caused by cancer and cancer treatment provides therapeutic opportunities to leverage the critical balance between DNA injury and repair. Blocking repair and pushing damaged DNA through the cell cycle using therapeutic inhibitors exemplify the 'pushmi-pullyu' effect of disrupted DNA repair. DNA repair inhibitors (DNARi) can be separated into five biofunctional categories: sensors, mediators, transducers, effectors, and collaborators that recognize DNA damage, propagate injury DNA messages, regulate cell cycle checkpoints, and alter the microenvironment. The result is cancer therapeutics that takes advantage of clinical synthetic lethality, resulting in selective tumor cell kill. Here, we review recent considerations related to DNA repair and new DNARi agents and organize those findings to address future directions and clinical opportunities.
Keywords: DNA damage repair; DNA damage response; genomic instability; homologous recombination defects; mutational burden; synthetic lethality.
Published by Elsevier Inc.
Figures
References
-
- Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nature reviews Cancer. 2005;5:689–698. - PubMed
-
- McLornan DP, et al. Applying synthetic lethality for the selective targeting of cancer. The New England journal of medicine. 2014;371:1725–1735. - PubMed
-
- Dietlein F, et al. Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends in genetics : TIG. 2014;30:326–339. - PubMed
-
- Lindahl T, et al. Post-translational modification of poly(ADP-ribose) polymerase induced by DNA strand breaks. Trends Biochem Sci. 1995;20:405–411. - PubMed
-
- O’Connor MJ. Targeting the DNA Damage Response in Cancer. Molecular cell. 2015;60:547–560. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
