

Evaluation of exome variants using the Ion Proton Platform to sequence error-prone regions
- PMID: 28742110
- PMCID: PMC5524428
- DOI: 10.1371/journal.pone.0181304
Evaluation of exome variants using the Ion Proton Platform to sequence error-prone regions
Abstract
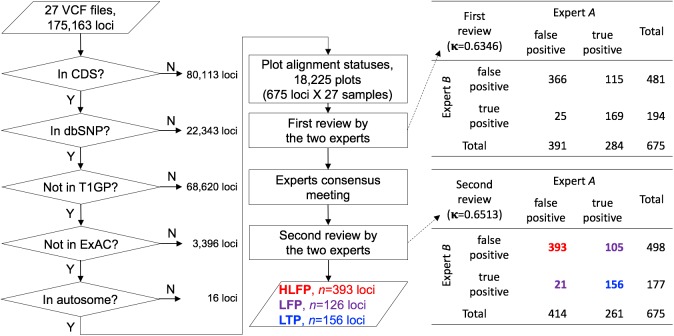
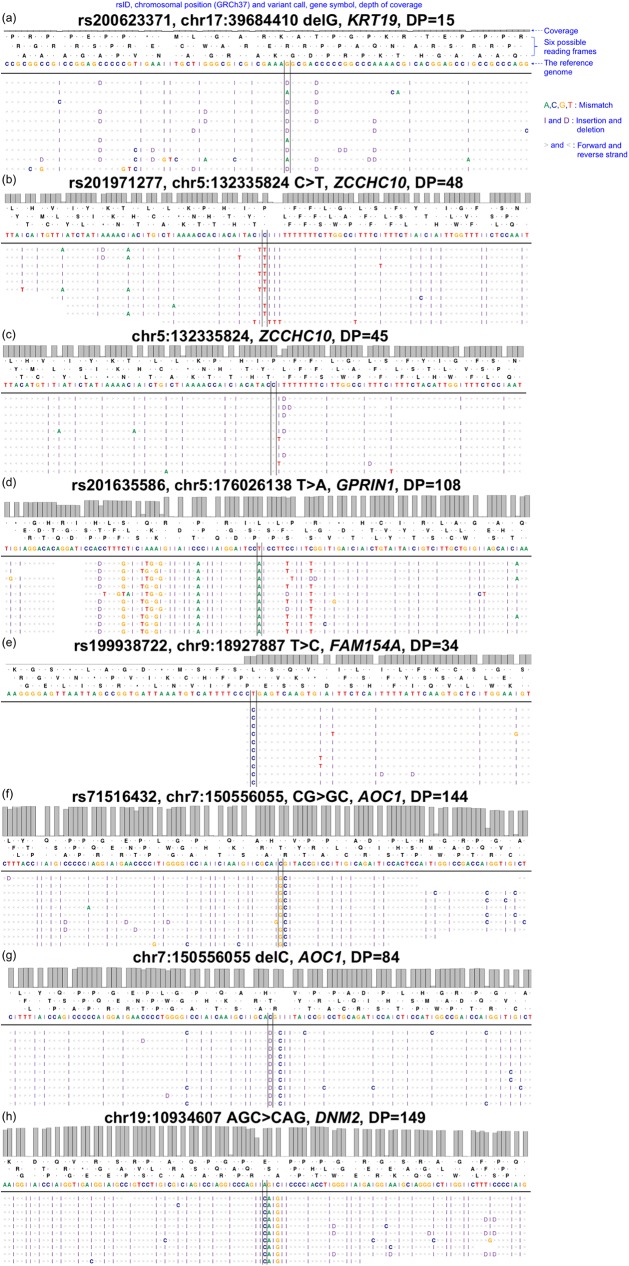
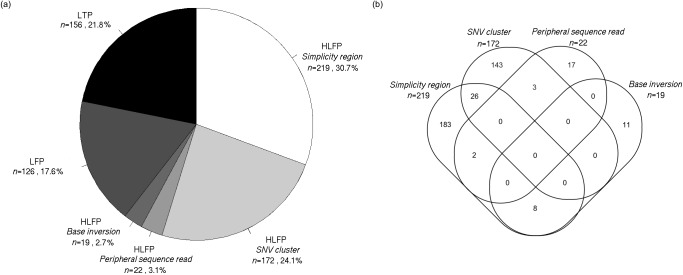
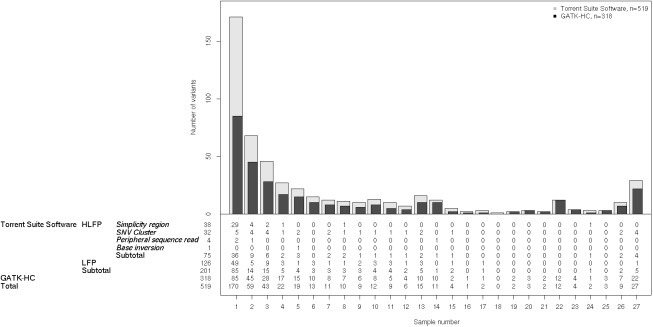
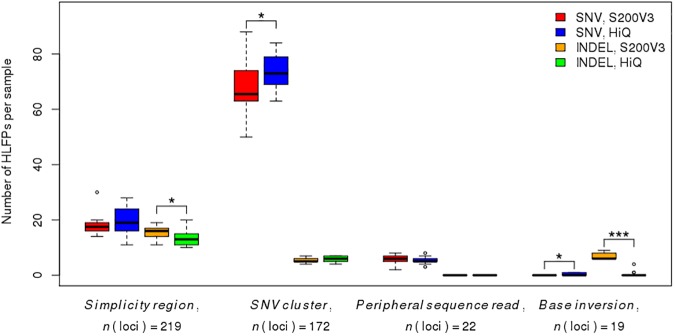
The Ion Proton sequencer from Thermo Fisher accurately determines sequence variants from target regions with a rapid turnaround time at a low cost. However, misleading variant-calling errors can occur. We performed a systematic evaluation and manual curation of read-level alignments for the 675 ultrarare variants reported by the Ion Proton sequencer from 27 whole-exome sequencing data but that are not present in either the 1000 Genomes Project and the Exome Aggregation Consortium. We classified positive variant calls into 393 highly likely false positives, 126 likely false positives, and 156 likely true positives, which comprised 58.2%, 18.7%, and 23.1% of the variants, respectively. We identified four distinct error patterns of variant calling that may be bioinformatically corrected when using different strategies: simplicity region, SNV cluster, peripheral sequence read, and base inversion. Local de novo assembly successfully corrected 201 (38.7%) of the 519 highly likely or likely false positives. We also demonstrate that the two sequencing kits from Thermo Fisher (the Ion PI Sequencing 200 kit V3 and the Ion PI Hi-Q kit) exhibit different error profiles across different error types. A refined calling algorithm with better polymerase may improve the performance of the Ion Proton sequencing platform.
Conflict of interest statement
Figures
Similar articles
-
Comparison and evaluation of two exome capture kits and sequencing platforms for variant calling.BMC Genomics. 2015 Aug 5;16(1):581. doi: 10.1186/s12864-015-1796-6. BMC Genomics. 2015. PMID: 26242175 Free PMC article.
-
Optimized detection of insertions/deletions (INDELs) in whole-exome sequencing data.PLoS One. 2017 Aug 9;12(8):e0182272. doi: 10.1371/journal.pone.0182272. eCollection 2017. PLoS One. 2017. PMID: 28792971 Free PMC article.
-
Calling known variants and identifying new variants while rapidly aligning sequence data.J Dairy Sci. 2019 Apr;102(4):3216-3229. doi: 10.3168/jds.2018-15172. Epub 2019 Feb 14. J Dairy Sci. 2019. PMID: 30772032
-
Exome versus transcriptome sequencing in identifying coding region variants.Expert Rev Mol Diagn. 2012 Apr;12(3):241-51. doi: 10.1586/erm.12.10. Expert Rev Mol Diagn. 2012. PMID: 22468815 Review.
-
Toward better understanding of artifacts in variant calling from high-coverage samples.Bioinformatics. 2014 Oct 15;30(20):2843-51. doi: 10.1093/bioinformatics/btu356. Epub 2014 Jun 27. Bioinformatics. 2014. PMID: 24974202 Free PMC article. Review.
Cited by
-
Predicting high confidence ctDNA somatic variants with ensemble machine learning models.Sci Rep. 2025 May 26;15(1):18384. doi: 10.1038/s41598-025-01326-2. Sci Rep. 2025. PMID: 40419568 Free PMC article.
-
Diagnostic Yield and Treatment Impact of Targeted Exome Sequencing in Early-Onset Epilepsy.Front Neurol. 2019 May 21;10:434. doi: 10.3389/fneur.2019.00434. eCollection 2019. Front Neurol. 2019. PMID: 31164858 Free PMC article.
-
Sharing genetic variants with the NGS pipeline is essential for effective genomic data sharing and reproducibility in health information exchange.Sci Rep. 2021 Jan 26;11(1):2268. doi: 10.1038/s41598-021-82006-9. Sci Rep. 2021. PMID: 33500538 Free PMC article.
References
-
- Saudi Mendeliome Group. Comprehensive gene panels provide advantages over clinical exome sequencing for Mendelian diseases. Genome Biol. BioMed Central; 2015;16: 134 doi: 10.1186/s13059-015-0693-2 - DOI - PMC - PubMed
-
- Ravenscroft G, Nolent F, Rajagopalan S, Meireles AM, Paavola KJ, Gaillard D, et al. Mutations of GPR126 are responsible for severe arthrogryposis multiplex congenita. Am J Hum Genet. 2015;96: 955–961. doi: 10.1016/j.ajhg.2015.04.014 - DOI - PMC - PubMed
-
- Begemann M, Zirn B, Santen G, Wirthgen E, Soellner L, Büttel H-M, et al. Paternally Inherited IGF2 Mutation and Growth Restriction. N Engl J Med. Massachusetts Medical Society; 2015;373: 349–356. doi: 10.1056/NEJMoa1415227 - DOI - PubMed
-
- Damiati E, Borsani G, Giacopuzzi E. Amplicon-based semiconductor sequencing of human exomes: performance evaluation and optimization strategies. Hum Genet. Springer Berlin Heidelberg; 2016;135: 499–511. doi: 10.1007/s00439-016-1656-8 - DOI - PMC - PubMed
-
- Quail M, Smith ME, Coupland P, Otto TD, Harris SR, Connor TR, et al. A tale of three next generation sequencing platforms: comparison of Ion torrent, pacific biosciences and illumina MiSeq sequencers. BMC Genomics. BioMed Central; 2012;13: 341 doi: 10.1186/1471-2164-13-341 - DOI - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
