

Capturing protein communities by structural proteomics in a thermophilic eukaryote
- PMID: 28743795
- PMCID: PMC5527848
- DOI: 10.15252/msb.20167412
Capturing protein communities by structural proteomics in a thermophilic eukaryote
Abstract
The arrangement of proteins into complexes is a key organizational principle for many cellular functions. Although the topology of many complexes has been systematically analyzed in isolation, their molecular sociology in situ remains elusive. Here, we show that crude cellular extracts of a eukaryotic thermophile, Chaetomium thermophilum, retain basic principles of cellular organization. Using a structural proteomics approach, we simultaneously characterized the abundance, interactions, and structure of a third of the C. thermophilum proteome within these extracts. We identified 27 distinct protein communities that include 108 interconnected complexes, which dynamically associate with each other and functionally benefit from being in close proximity in the cell. Furthermore, we investigated the structure of fatty acid synthase within these extracts by cryoEM and this revealed multiple, flexible states of the enzyme in adaptation to its association with other complexes, thus exemplifying the need for in situ studies. As the components of the captured protein communities are known-at both the protein and complex levels-this study constitutes another step forward toward a molecular understanding of subcellular organization.
Keywords: computational modeling; cryo‐electron microscopy; fatty acid synthase; interaction proteomics; metabolon.
© 2017 European Molecular Biology Laboratory. Published under the terms of the CC BY 4.0 license.
Figures

We combined computational modeling approaches adapted from network biology (molecular profiling) with molecular biophysics, electron microscopy (EM; structural profiling), and quantitative and cross‐linking mass spectrometry (interface profiling) to systematically chart and characterize the organization of protein complexes into functional, local communities. Large‐scale electron microscopy and cross‐linking mass spectrometry are used as validation tools.
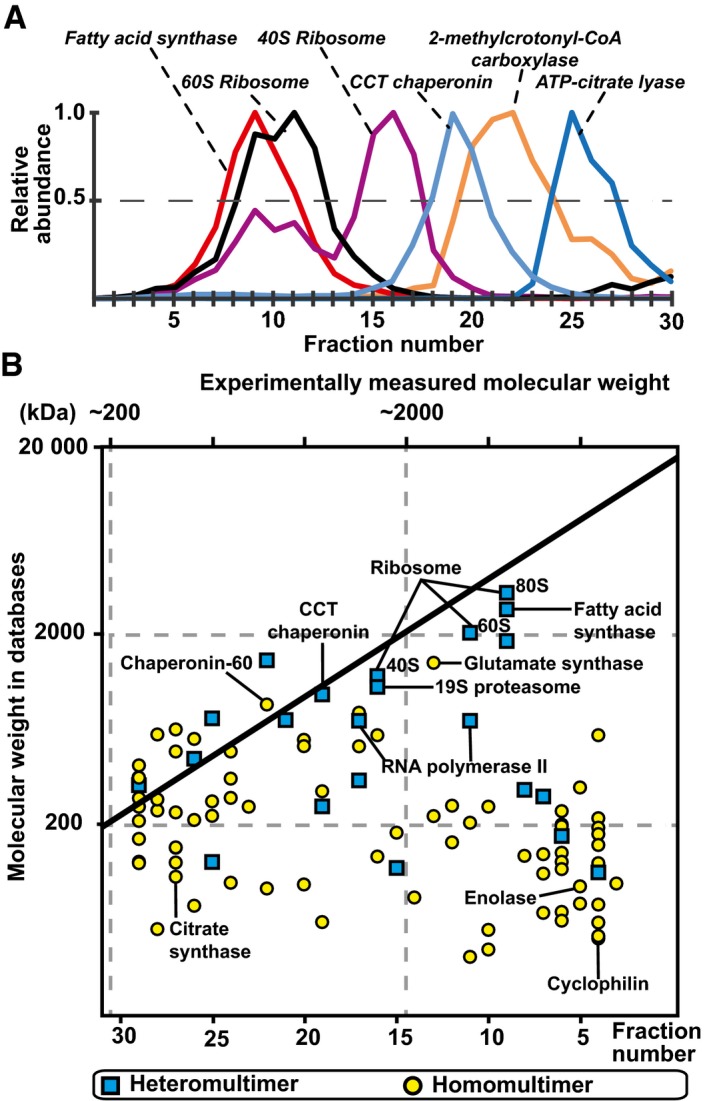
Elution of selected protein complexes as a function of their retention times (see Appendix Fig S4 for their corresponding subunit elutions).
Scatter plot indicating discrepancies in the expected and measured molecular weights of 102 protein complexes that elute as a single peak; 50% of protein complexes are observed to have higher molecular weights than structurally characterized, indicating that they are organized in higher‐order assemblies.
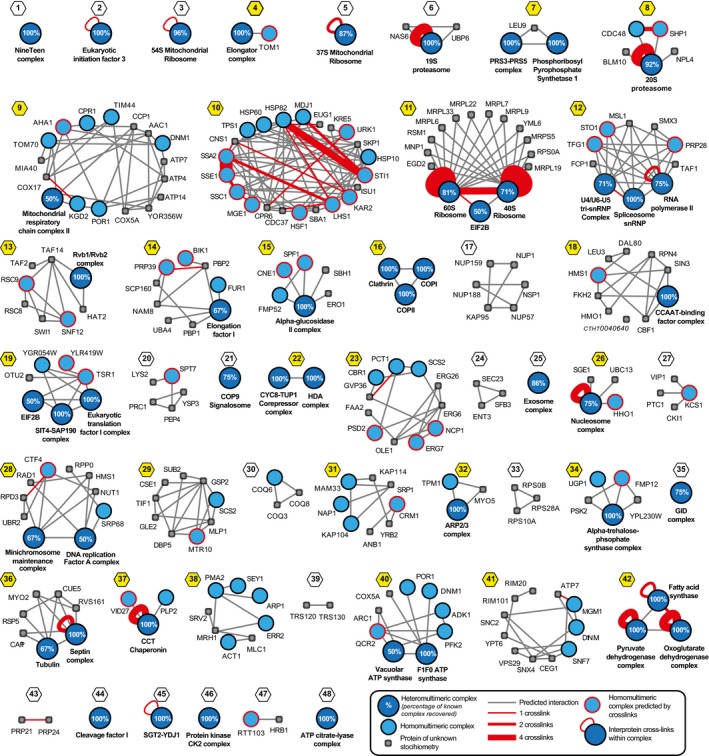
Integration of experimental elution data, known functional associations, and predicted interaction interfaces from homologous proteins allow the creation of a high‐quality network with interconnected protein complexes (Appendix Figs S8–S10). Here, known protein complexes are shown in blue and other physically associated proteins in gray, predicted interactions of complexes as gray lines and cross‐links as red lines, and cross‐links between different subunits of a heteromultimeric complex are represented with red loops (see insert). Communities containing multiple complexes are highlighted with yellow; numbering and naming of complexes and communities are described in the legend of Appendix Fig S9.
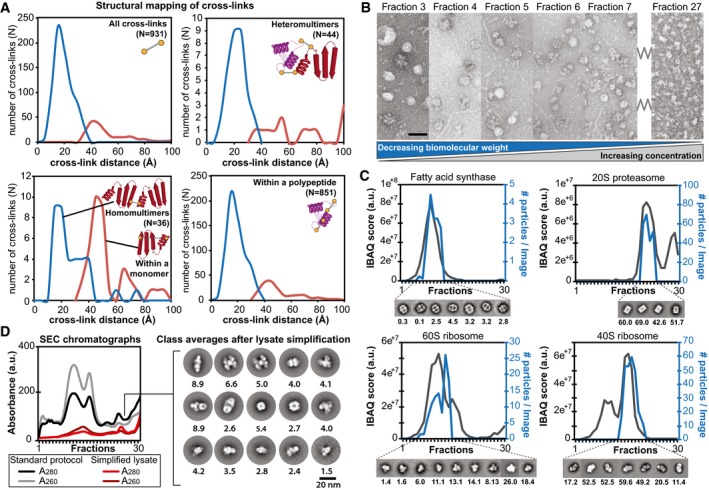
Distance distributions of identified cross‐links on top of the modeled protein complexes and identification of novel interactions. Satisfied distances are shown in blue and over‐length cross‐links are shown in red.
Negatively stained electron micrographs of fractions 3–7 and 27 directly derived from size exclusion chromatography showing the structural signatures and their structural integrity within the fractions. Decreasing molecular weight correlates with increased protein concentration as a function of protein complex elution is highlighted. Scale bar: 60 nm.
Abundance profiles as determined by quantitative mass spectrometry correlate with the number of observed single particles of the corresponding structural signature within the negative‐staining electron micrographs; shown for fatty acid synthase, 20S proteasome, 60S and 40S ribosome (the number of particles per image per fraction is indicated below the class averages).
Simplification of lysate (collecting only the flow‐through from anion exchange chromatography) prior to SEC separation allows class averaging of structural signatures from complex fractions that were previously too low abundant. The number of particles per image per fraction is indicated below the class averages.
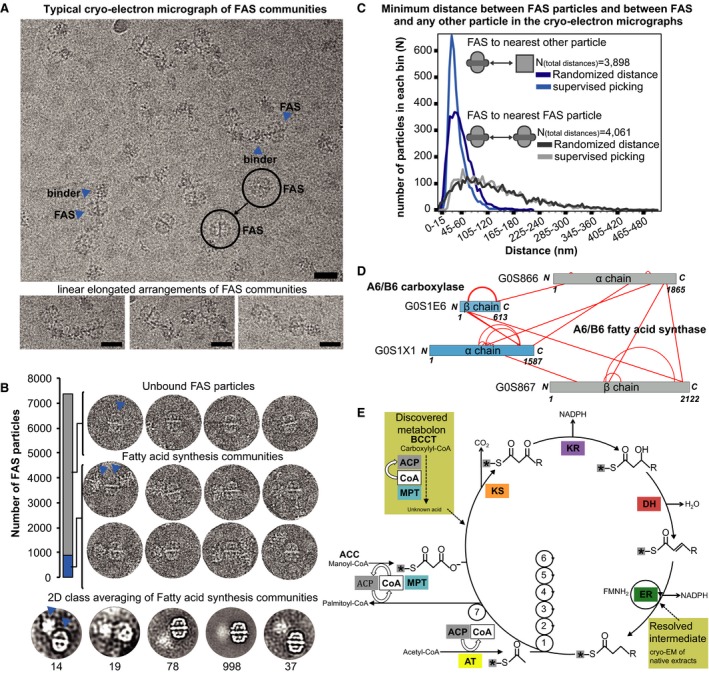
Communities in fatty acid metabolism and the quantification of intra‐community distances within cryo‐electron micrographs. Fatty acid synthase (FAS) frequently interacts with other sizeable protein complexes in a linear “pearl‐string‐like” arrangement and usually localizes at the edges of the community. Scale bars correspond to 25 nm. FAS particles (circles) and their nearest neighbors (arrow heads) are indicated.
Additional density outside of the ctFAS dome is observed in ∼10% of the single particles; 2D class averages shown at the bottom. The arrow heads show typical assemblies within the pool of particles. In 90% of all cases, isolated FAS particles are seen (unbound state). In the remaining 10%, higher‐order protein assemblies comprised of FAS particles and high molecular weight binders are seen (bound state).
Related to (A). Calculation of minimum distances between pairs of FAS molecules as well as FAS molecules and their closest non‐FAS neighbors in comparison with random distributions. Whereas FAS molecules are randomly distributed, their binders are not, confirming physical interactions. Supervised picking means that all single particles were manually picked from the images. Randomized distance means that these manually picked particles were assigned random coordinates in each image (randomization of x, y coordinates considering image borders) and then their distance is calculated.
Cross‐linking mass spectrometry data show that the binder is a carboxylase that is bound to the malonyl transacylase domain and acyl carrier protein (ACP) is in the vicinity, considering cross‐link length and the positions of the lysine on the ctFAS structure. Cross‐links come from both affinity‐purified and fractionated cell extracts.
The molecular mechanisms in fatty acid synthesis (Wakil et al, 1983), and the relevance of the position of the ACP (see Fig 6 for details) and carboxylase to the catalytic cycle is indicated (see text). ACP, acyl carrier protein; CoA, acetyl‐coenzyme A; MPT, malonyl/palmitoyl transferase; KS, ketoacyl synthase; KR, ketoacyl reductase; DH, dehydratase; ER, enoyl reductase; AT, acetyltransferase. Asterisks represent the acyl carrier protein (ACP).
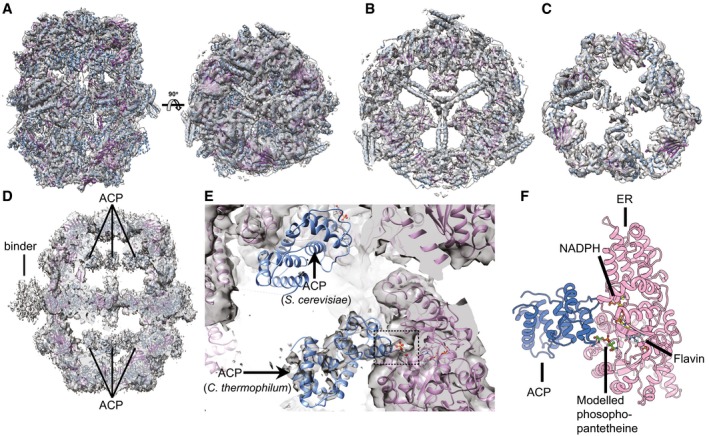
The cryoEM map of Chaetomium thermophilum fatty acid synthase (ctFAS) is shown isosurface rendered and superimposed with the fitted X‐ray structure of yeast FAS (Jenni et al, 2007). The domes and the cap show the unambiguous fit of α‐helices and β‐strands.
A slice through the central wheel of fungal FAS. The pitch of α‐helices is resolved.
As for (B) but sliced through the dome structure.
Location of acyl carrier protein (ACP) within the cryoEM map of ctFAS and the position of additional density outside the dome.
Fit of ACP in the cryoEM map of ctFAS and comparison with the crystallographically determined location in yeast FAS; additional density within the active site possibly resembling the acyl chain bound on the ACP is observed.
Molecular model of the interaction between the ACP and the ER domains of ctFAS in cartoon representation. The model was derived from a rigid fit from (B) and subsequently flexibly refined for clash removal and interface energetics optimization.
References
-
- Amlacher S, Sarges P, Flemming D, van Noort V, Kunze R, Devos DP, Arumugam M, Bork P, Hurt E (2011) Insight into structure and assembly of the nuclear pore complex by utilizing the genome of a eukaryotic thermophile. Cell 146: 277–289 - PubMed
-
- von Appen A, Kosinski J, Sparks L, Ori A, DiGuilio AL, Vollmer B, Mackmull MT, Banterle N, Parca L, Kastritis P, Buczak K, Mosalaganti S, Hagen W, Andres‐Pons A, Lemke EA, Bork P, Antonin W, Glavy JS, Bui KH, Beck M (2015) In situ structural analysis of the human nuclear pore complex. Nature 526: 140–143 - PMC - PubMed
-
- Barabasi AL, Oltvai ZN (2004) Network biology: understanding the cell's functional organization. Nat Rev Genet 5: 101–113 - PubMed
-
- Beck M, Baumeister W (2016) Cryo‐electron tomography: can it reveal the molecular sociology of cells in atomic detail? Trends Cell Biol 26: 825–837 - PubMed
-
- Benschop JJ, Brabers N, van Leenen D, Bakker LV, van Deutekom HW, van Berkum NL, Apweiler E, Lijnzaad P, Holstege FC, Kemmeren P (2010) A consensus of core protein complex compositions for Saccharomyces cerevisiae . Mol Cell 38: 916–928 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
