

A Landscape of Therapeutic Cooperativity in KRAS Mutant Cancers Reveals Principles for Controlling Tumor Evolution
- PMID: 28746882
- PMCID: PMC5567854
- DOI: 10.1016/j.celrep.2017.07.006
A Landscape of Therapeutic Cooperativity in KRAS Mutant Cancers Reveals Principles for Controlling Tumor Evolution
Abstract
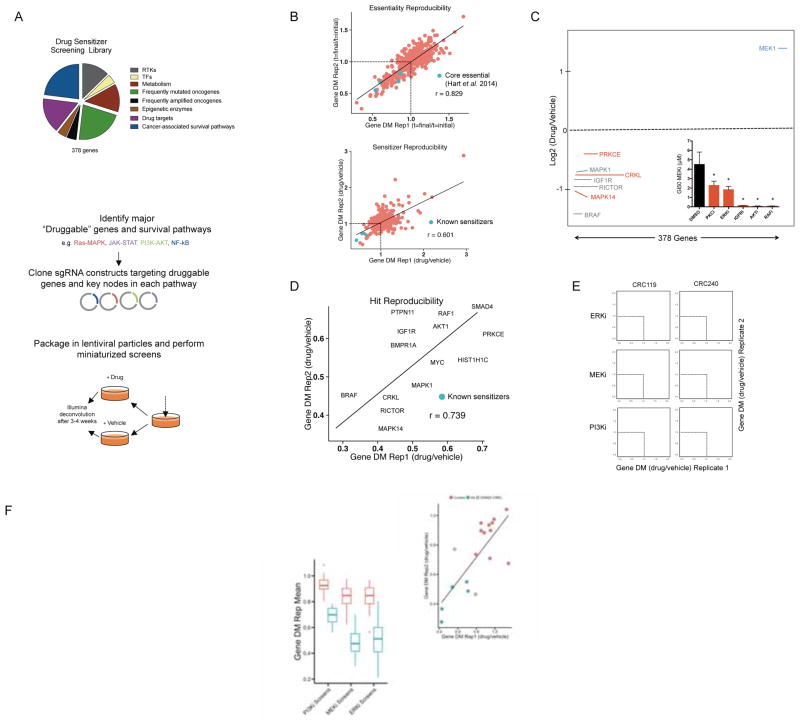
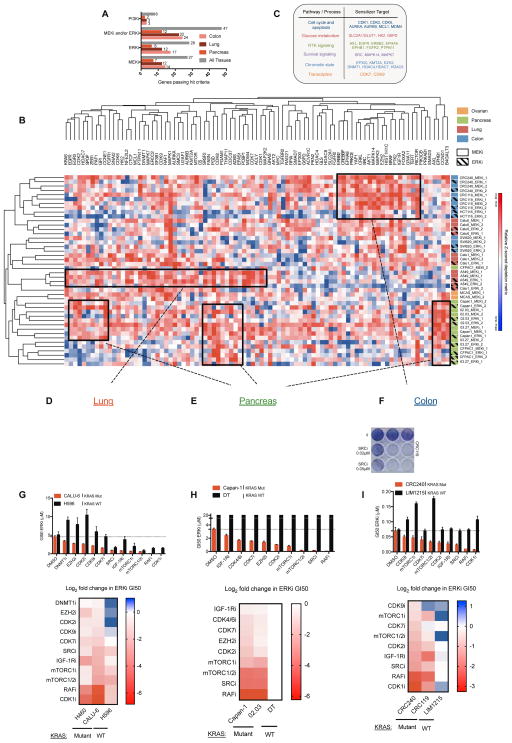
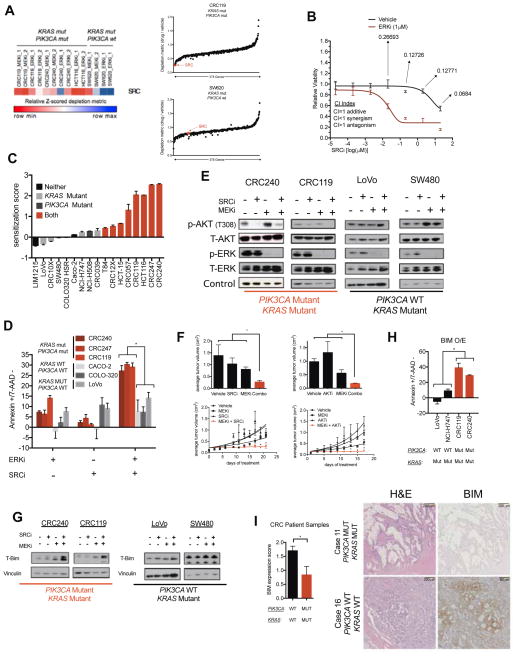
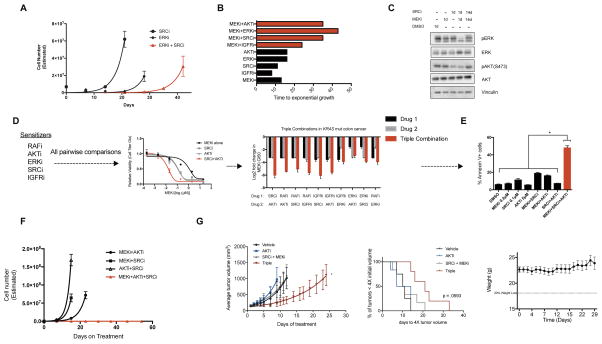
Combinatorial inhibition of effector and feedback pathways is a promising treatment strategy for KRAS mutant cancers. However, the particular pathways that should be targeted to optimize therapeutic responses are unclear. Using CRISPR/Cas9, we systematically mapped the pathways whose inhibition cooperates with drugs targeting the KRAS effectors MEK, ERK, and PI3K. By performing 70 screens in models of KRAS mutant colorectal, lung, ovarian, and pancreas cancers, we uncovered universal and tissue-specific sensitizing combinations involving inhibitors of cell cycle, metabolism, growth signaling, chromatin regulation, and transcription. Furthermore, these screens revealed secondary genetic modifiers of sensitivity, yielding a SRC inhibitor-based combination therapy for KRAS/PIK3CA double-mutant colorectal cancers (CRCs) with clinical potential. Surprisingly, acquired resistance to combinations of growth signaling pathway inhibitors develops rapidly following treatment, but by targeting signaling feedback or apoptotic priming, it is possible to construct three-drug combinations that greatly delay its emergence.
Keywords: BIM; CRISPR/Cas9; KRAS; PIK3CA; SRC; apoptosis; drug resistance; pooled screening; synthetic lethality.
Copyright © 2017 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
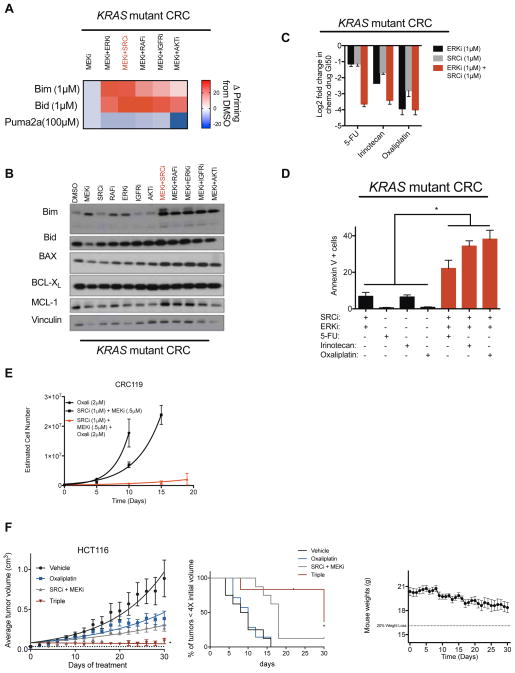
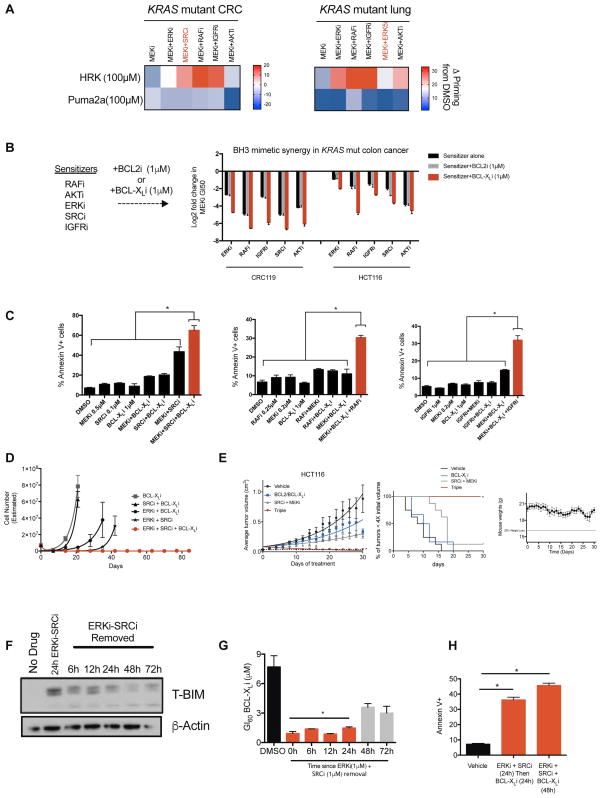
References
Publication types
MeSH terms
Substances
Grants and funding
- K00 CA222728/CA/NCI NIH HHS/United States
- F99 CA222728/CA/NCI NIH HHS/United States
- F30 CA206348/CA/NCI NIH HHS/United States
- F32 CA180569/CA/NCI NIH HHS/United States
- P30 CA016086/CA/NCI NIH HHS/United States
- U01 CA199235/CA/NCI NIH HHS/United States
- UL1 TR001117/TR/NCATS NIH HHS/United States
- T32 GM007171/GM/NIGMS NIH HHS/United States
- K12 HD043446/HD/NICHD NIH HHS/United States
- F32 CA206234/CA/NCI NIH HHS/United States
- T32 GM007754/GM/NIGMS NIH HHS/United States
- F31 CA195967/CA/NCI NIH HHS/United States
- F32 CA134171/CA/NCI NIH HHS/United States
- R01 CA123031/CA/NCI NIH HHS/United States
- R01 CA207083/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
