

Unbiased whole-genome deep sequencing of human and porcine stool samples reveals circulation of multiple groups of rotaviruses and a putative zoonotic infection
- PMID: 28748110
- PMCID: PMC5522372
- DOI: 10.1093/ve/vew027
Unbiased whole-genome deep sequencing of human and porcine stool samples reveals circulation of multiple groups of rotaviruses and a putative zoonotic infection
Abstract
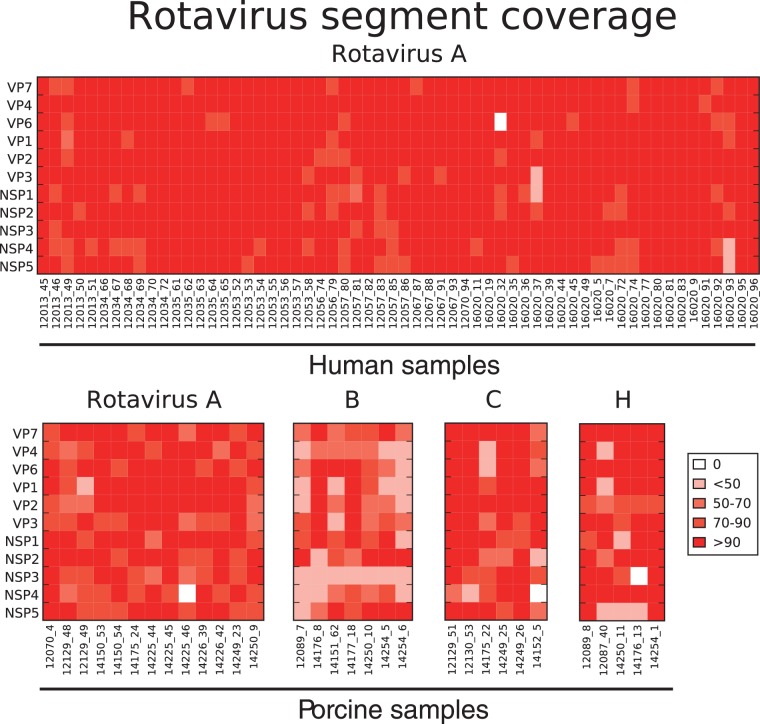
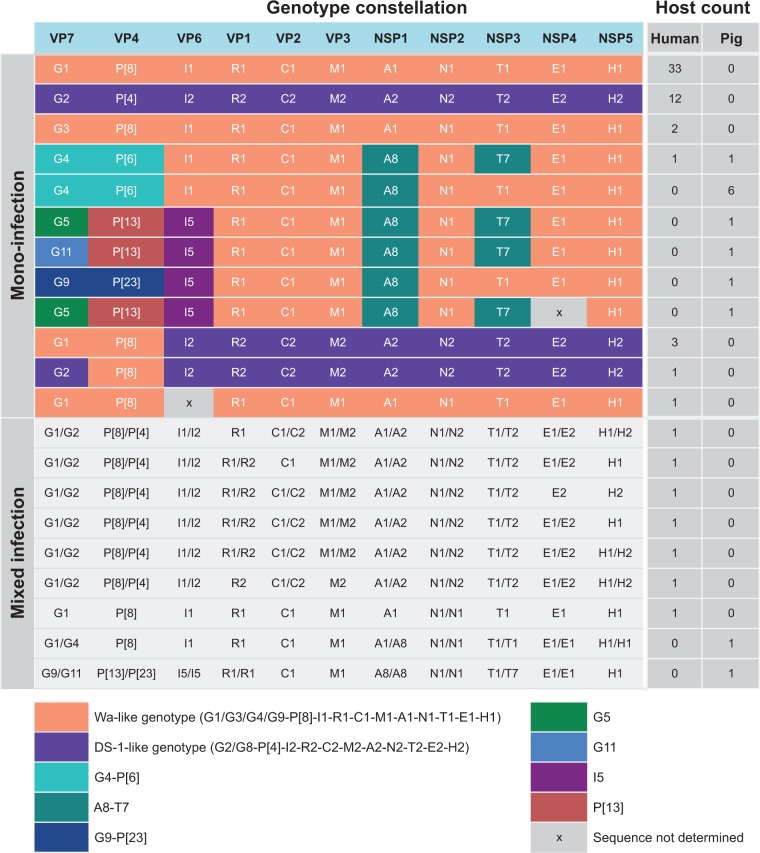
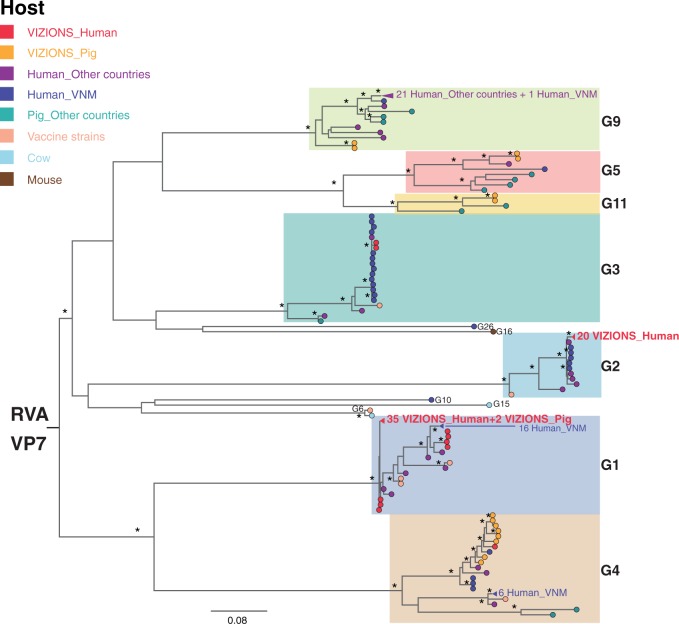
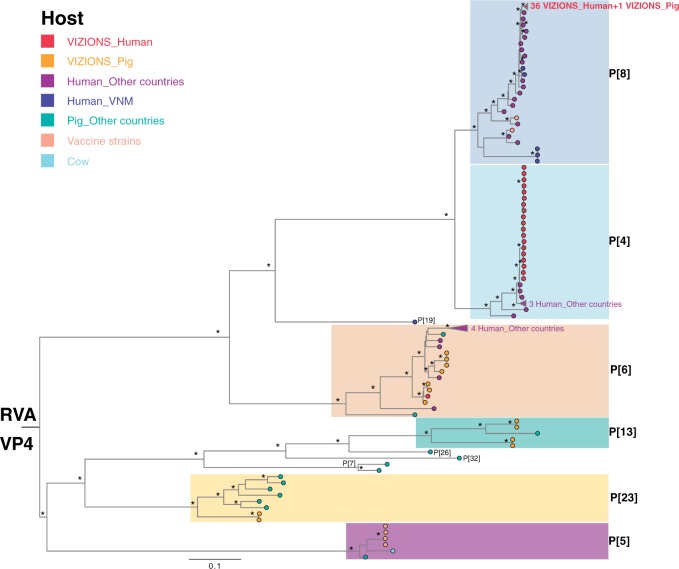
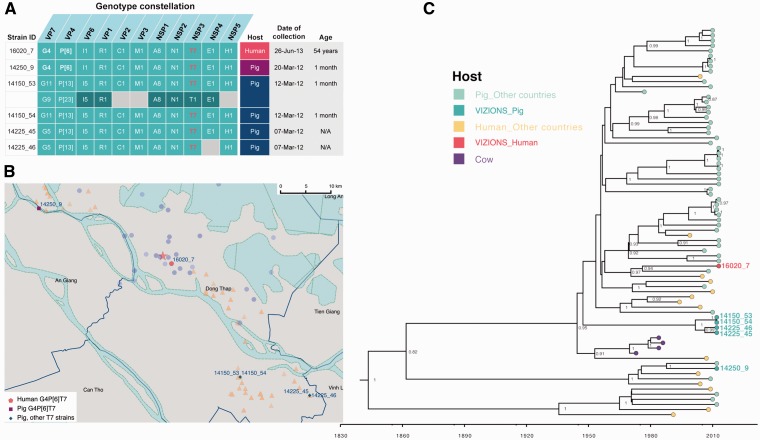
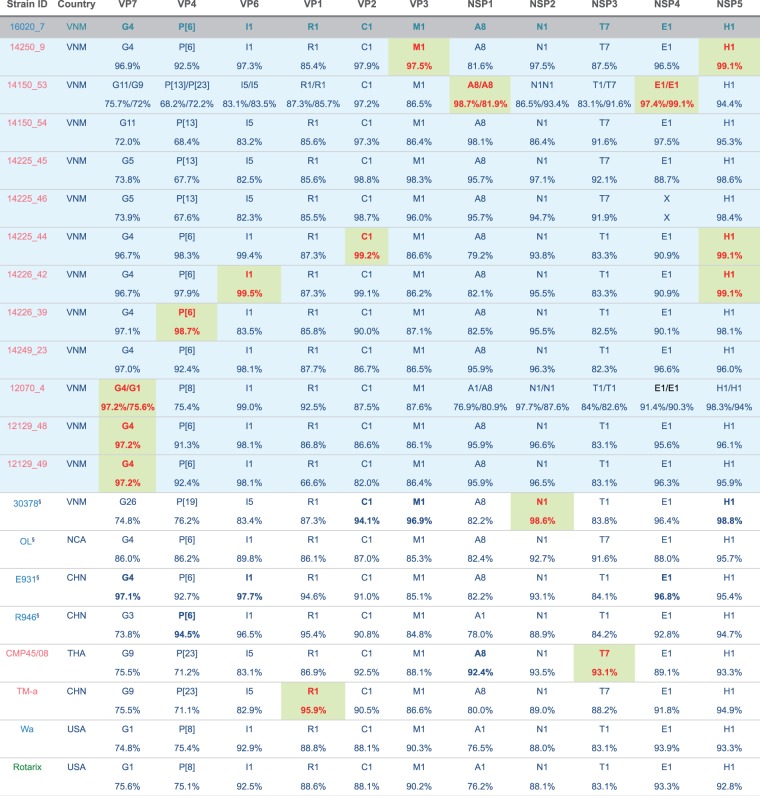
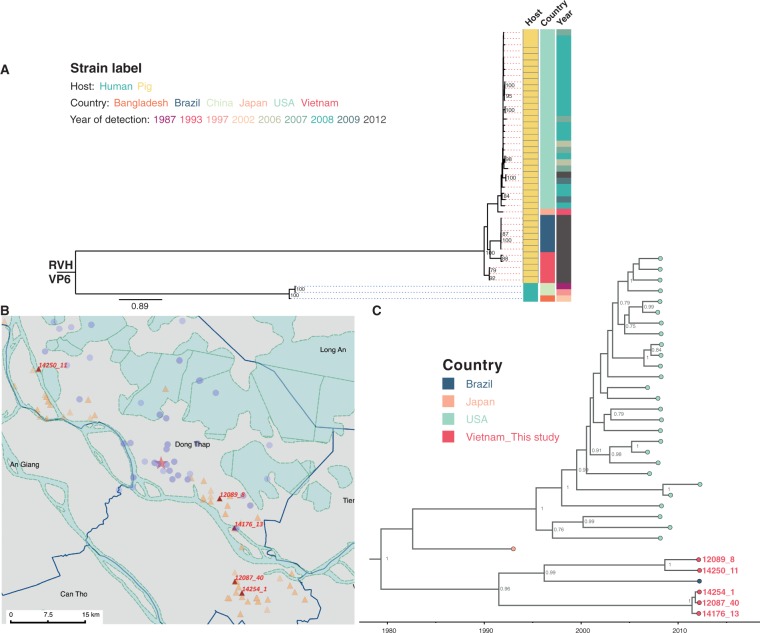
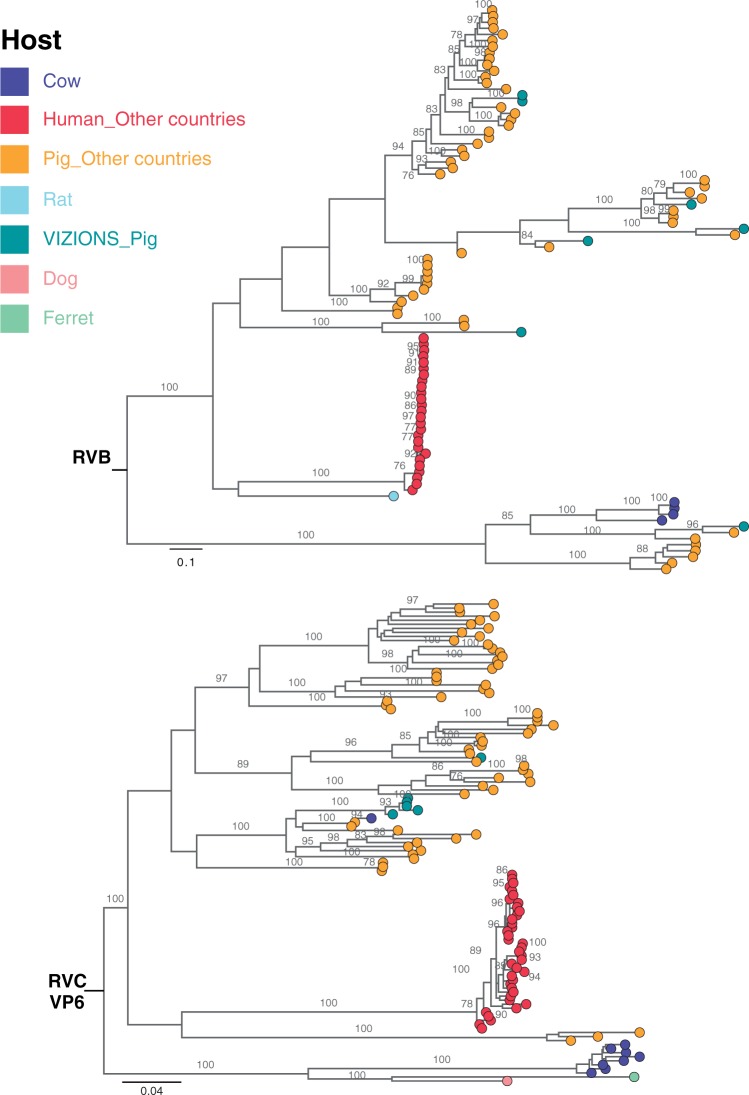
Coordinated and synchronous surveillance for zoonotic viruses in both human clinical cases and animal reservoirs provides an opportunity to identify interspecies virus movement. Rotavirus (RV) is an important cause of viral gastroenteritis in humans and animals. In this study, we document the RV diversity within co-located humans and animals sampled from the Mekong delta region of Vietnam using a primer-independent, agnostic, deep sequencing approach. A total of 296 stool samples (146 from diarrhoeal human patients and 150 from pigs living in the same geographical region) were directly sequenced, generating the genomic sequences of sixty human rotaviruses (all group A) and thirty-one porcine rotaviruses (thirteen group A, seven group B, six group C, and five group H). Phylogenetic analyses showed the co-circulation of multiple distinct RV group A (RVA) genotypes/strains, many of which were divergent from the strain components of licensed RVA vaccines, as well as considerable virus diversity in pigs including full genomes of rotaviruses in groups B, C, and H, none of which have been previously reported in Vietnam. Furthermore, the detection of an atypical RVA genotype constellation (G4-P[6]-I1-R1-C1-M1-A8-N1-T7-E1-H1) in a human patient and a pig from the same region provides some evidence for a zoonotic event.
Keywords: deep sequencing; rotavirus; virus surveillance; whole genomes; zoonotic infection.
Figures
Similar articles
-
Metagenomic sequencing generates the whole genomes of porcine rotavirus A, C, and H from the United States.PLoS One. 2020 Dec 29;15(12):e0244498. doi: 10.1371/journal.pone.0244498. eCollection 2020. PLoS One. 2020. PMID: 33373390 Free PMC article.
-
Genomic characterization of G3P[6], G4P[6] and G4P[8] human rotaviruses from Wuhan, China: Evidence for interspecies transmission and reassortment events.Infect Genet Evol. 2015 Jul;33:55-71. doi: 10.1016/j.meegid.2015.04.010. Epub 2015 Apr 16. Infect Genet Evol. 2015. PMID: 25891280
-
Complete genome characterization by nanopore sequencing of rotaviruses A, B, and C circulating on large-scale pig farms in Russia.Virol J. 2024 Nov 13;21(1):289. doi: 10.1186/s12985-024-02567-9. Virol J. 2024. PMID: 39538316 Free PMC article.
-
Detection and genetic characterization of porcine group A rotaviruses in asymptomatic pigs in smallholder farms in East Africa: predominance of P[8] genotype resembling human strains.Vet Microbiol. 2015 Feb 25;175(2-4):195-210. doi: 10.1016/j.vetmic.2014.11.027. Epub 2014 Dec 10. Vet Microbiol. 2015. PMID: 25541378
-
Prevalence and genomic characterization of rotavirus group A genotypes in piglets from southern highlands and eastern Tanzania.Heliyon. 2022 Nov 23;8(11):e11750. doi: 10.1016/j.heliyon.2022.e11750. eCollection 2022 Nov. Heliyon. 2022. PMID: 36468104 Free PMC article.
Cited by
-
In Vitro Inactivation of Respiratory Viruses and Rotavirus by the Oral Probiotic Strain Weissella cibaria CMS1.Probiotics Antimicrob Proteins. 2022 Aug;14(4):760-766. doi: 10.1007/s12602-022-09947-z. Epub 2022 May 10. Probiotics Antimicrob Proteins. 2022. PMID: 35536505 Free PMC article.
-
Extended analyses of rotavirus C (RVC) G-types and P-types reveal new cut-off value for the G-types and reclassification of strains.J Virol. 2025 May 20;99(5):e0004925. doi: 10.1128/jvi.00049-25. Epub 2025 Apr 15. J Virol. 2025. PMID: 40231817 Free PMC article.
-
Sample descriptors linked to metagenomic sequencing data from human and animal enteric samples from Vietnam.Sci Data. 2019 Oct 15;6(1):202. doi: 10.1038/s41597-019-0215-2. Sci Data. 2019. PMID: 31615980 Free PMC article.
-
Identification of a potential interspecies reassortant rotavirus G and avastrovirus 2 co-infection from black-headed gull (Chroicocephalus ridibundus) in Hungary.PLoS One. 2025 Mar 24;20(3):e0317400. doi: 10.1371/journal.pone.0317400. eCollection 2025. PLoS One. 2025. PMID: 40127066 Free PMC article.
-
Frequent Occurrence of Simultaneous Infection with Multiple Rotaviruses in Swiss Pigs.Viruses. 2022 May 23;14(5):1117. doi: 10.3390/v14051117. Viruses. 2022. PMID: 35632858 Free PMC article.
References
-
- Anh D. D., et al. (2006) ‘The Burden of Rotavirus Diarrhea in Khanh Hoa Province, Vietnam: Baseline Assessment for a Rotavirus Vaccine Trial’, Pediatrics Infectious Disease Journal, 25: 37–40. - PubMed
-
- Armah G. E., et al. (2010) ‘Efficacy of Pentavalent Rotavirus Vaccine Against Severe Rotavirus Gastroenteritis in Infants in Developing Countries in Sub-Saharan Africa: A Randomised, Double-Blind, Placebo-Controlled Trial’, Lancet, 376: 606–14. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
