

Utility of Genetic Testing for Confirmation of Abnormal Newborn Screening in Disorders of Long-Chain Fatty Acids: A Missed Case of Carnitine Palmitoyltransferase 1A (CPT1A) Deficiency
- PMID: 28748224
- PMCID: PMC5523953
- DOI: 10.3390/ijns3020010
Utility of Genetic Testing for Confirmation of Abnormal Newborn Screening in Disorders of Long-Chain Fatty Acids: A Missed Case of Carnitine Palmitoyltransferase 1A (CPT1A) Deficiency
Abstract
An 18-month-old male was evaluated after presenting with disproportionately elevated liver transaminases in the setting of acute gastroenteritis. He had marked hepatomegaly on physical exam that was later confirmed with an abdominal ultrasound. Given this clinical picture, suspicion for a fatty acid oxidation disorder was raised. Further investigation revealed that his initial newborn screen was positive for carnitine palmitoyltransferase 1A (CPT1A) deficiency-a rare autosomal recessive disorder of long-chain fatty acid oxidation. Confirmatory biochemical testing in the newborn period showed carnitine levels to be unexpectedly low with a normal acylcarnitine profile. Thus, it was considered to be a false-positive newborn screen and metabolic follow-up was not recommended. Repeat biochemical testing during this hospitalization revealed a normal acylcarnitine profile. The only abnormalities noted were a low proportion of acylcarnitine species from plasma, an elevated free-to-total carnitine ratio, and mild hypoketotic medium chain dicarboxylic aciduria on urine organic acids. Gene sequencing of CPT1A revealed a novel homozygous splice site variant that confirmed his diagnosis. CPT1A deficiency has a population founder effect in the Inuit and other Arctic groups, but has not been previously reported in persons of Ashkenazi Jewish ancestry.
Keywords: Ashkenazi Jewish; CPT1A; carnitine palmitoyltransferase deficiency; elevated liver transaminases; fatty acid oxidation disorders; neonatal screening.
Conflict of interest statement
Conflicts of Interest: The authors declare no conflicts of interest.
Figures
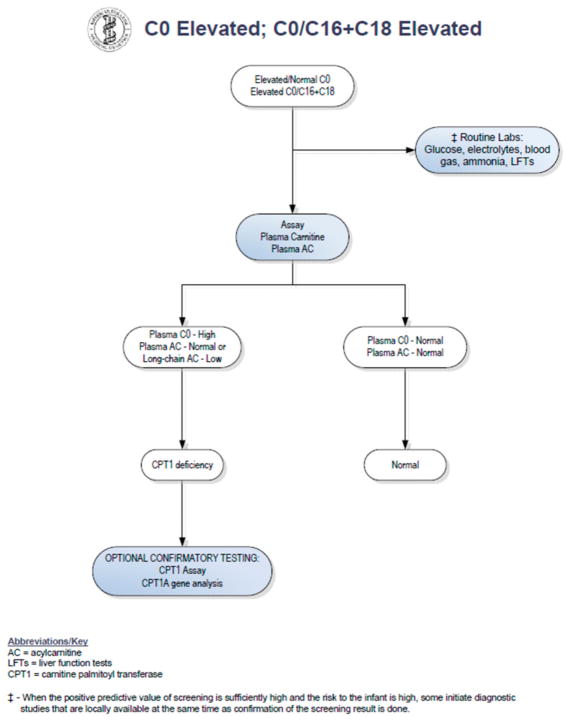
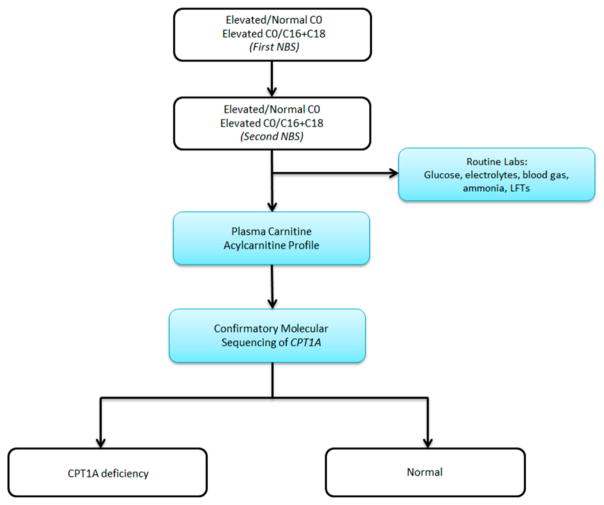
Similar articles
-
Carnitine Palmitoyltransferase 1A Deficiency.2005 Jul 27 [updated 2025 Feb 20]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2005 Jul 27 [updated 2025 Feb 20]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 20301700 Free Books & Documents. Review.
-
A patient with atypical presentation of chronic hepatosteatosis harboring a novel variant in the CPT1A gene.Eur J Med Genet. 2021 Jan;64(1):104034. doi: 10.1016/j.ejmg.2020.104034. Epub 2020 Aug 8. Eur J Med Genet. 2021. PMID: 32781271
-
Novel mutation in carnitine palmitoyltransferase 1A detected through newborn screening for a presymptomatic case in China: a case report.Ital J Pediatr. 2021 Jul 7;47(1):154. doi: 10.1186/s13052-021-01094-5. Ital J Pediatr. 2021. PMID: 34233743 Free PMC article.
-
Novel metabolic and molecular findings in hepatic carnitine palmitoyltransferase I deficiency.Mol Genet Metab. 2005 Nov;86(3):337-43. doi: 10.1016/j.ymgme.2005.07.022. Epub 2005 Sep 16. Mol Genet Metab. 2005. PMID: 16146704
-
Carnitine-acylcarnitine translocase deficiency, clinical, biochemical and genetic aspects.Mol Aspects Med. 2004 Oct-Dec;25(5-6):521-32. doi: 10.1016/j.mam.2004.06.007. Mol Aspects Med. 2004. PMID: 15363639 Review.
Cited by
-
Three Novel and One Potential Hotspot CPT1A Variants in Chinese Patients With Carnitine Palmitoyltransferase 1A Deficiency.Front Pediatr. 2021 Nov 12;9:771922. doi: 10.3389/fped.2021.771922. eCollection 2021. Front Pediatr. 2021. PMID: 34869124 Free PMC article.
-
Newborn Screening for Mitochondrial Carnitine-Acylcarnitine Cycle Disorders in Zhejiang Province, China.Front Genet. 2022 Mar 14;13:823687. doi: 10.3389/fgene.2022.823687. eCollection 2022. Front Genet. 2022. PMID: 35360862 Free PMC article.
-
Mitochondrial CPT1A: Insights into structure, function, and basis for drug development.Front Pharmacol. 2023 Mar 23;14:1160440. doi: 10.3389/fphar.2023.1160440. eCollection 2023. Front Pharmacol. 2023. PMID: 37033619 Free PMC article. Review.
-
Essential oils can cause false-positive results of medium-chain acyl-CoA dehydrogenase deficiency.Mol Genet Metab Rep. 2020 Nov 5;25:100674. doi: 10.1016/j.ymgmr.2020.100674. eCollection 2020 Dec. Mol Genet Metab Rep. 2020. PMID: 33204637 Free PMC article.
References
-
- Hughes I. In: Pediatric Endocrinology and Inborn Errors of Metabolism. 1. Sarafoglou K, Hoffman G, Roth K, editors. McGraw-Hill Education; New York, NY, USA: 2009.
-
- Korman SH, Waterham HR, Gutman A, Jakobs C, Wanders RJA. Novel metabolic and molecular findings in hepatic carnitine palmitoyltransferase I deficiency. Mol Gen Metab. 2005;86:337–343. - PubMed
-
- Tsuburaya R, Sakamoto O, Arai N, Kobayashi H, Hasegawa Y, Yamaguchi S, Tsuchiya S. Molecular analysis of a presymptomatic case of carnitine palmitoyl transferase I (CPTI) deficiency detected by tandem mass spectrometry newborn screening in Japan. Brain Dev. 2010;32:409–411. - PubMed
-
- Fingerhut R, Roschinger W, Muntau A, Roscher A. Hepatic carnitine palmitoyltransferase I deficiency: Acylcarnitine profiles in blood spots are highly specific. Clin Chem. 2010;47:1763–1768. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
