

Integrated time-lapse and single-cell transcription studies highlight the variable and dynamic nature of human hematopoietic cell fate commitment
- PMID: 28749943
- PMCID: PMC5531424
- DOI: 10.1371/journal.pbio.2001867
Integrated time-lapse and single-cell transcription studies highlight the variable and dynamic nature of human hematopoietic cell fate commitment
Abstract
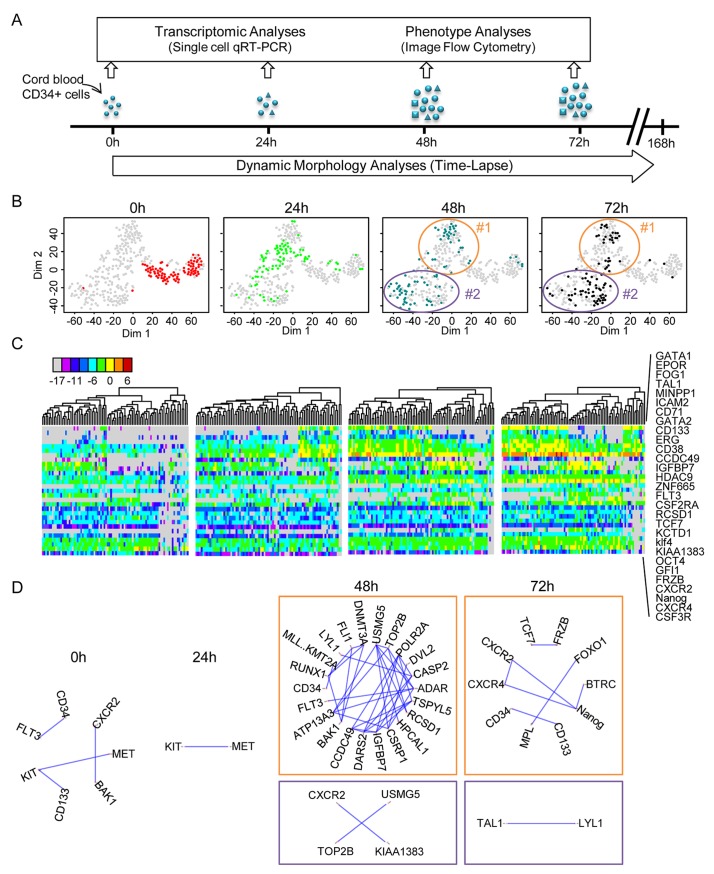
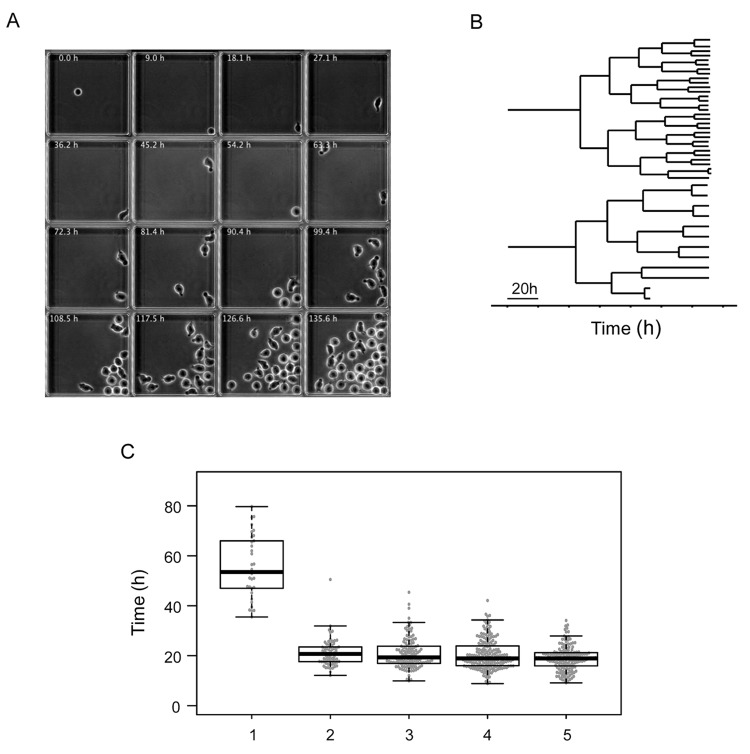
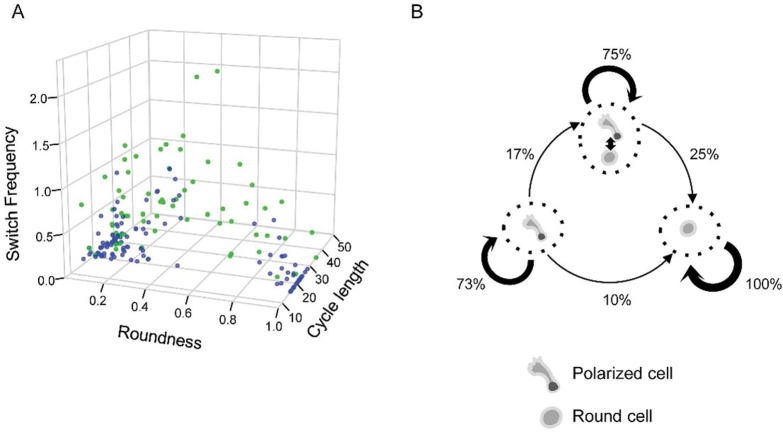
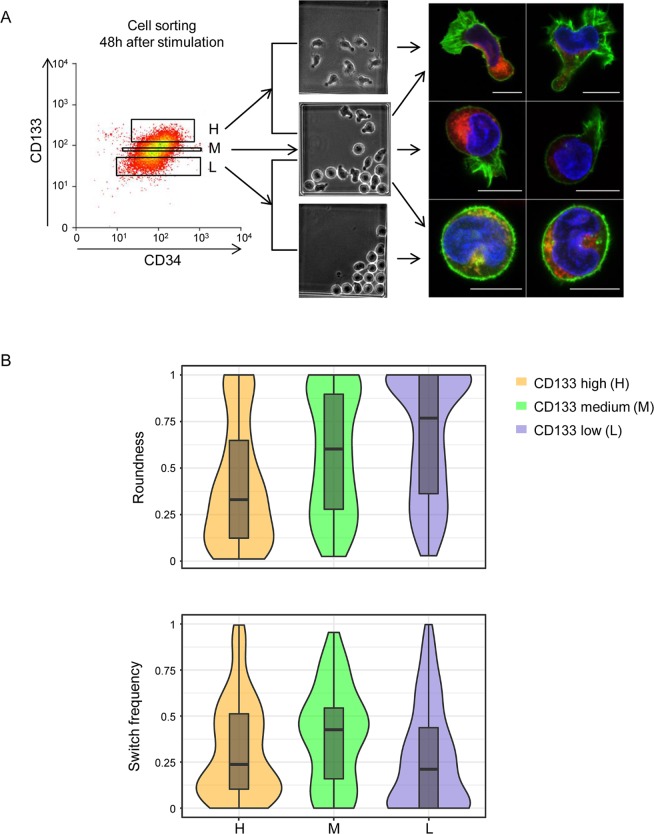
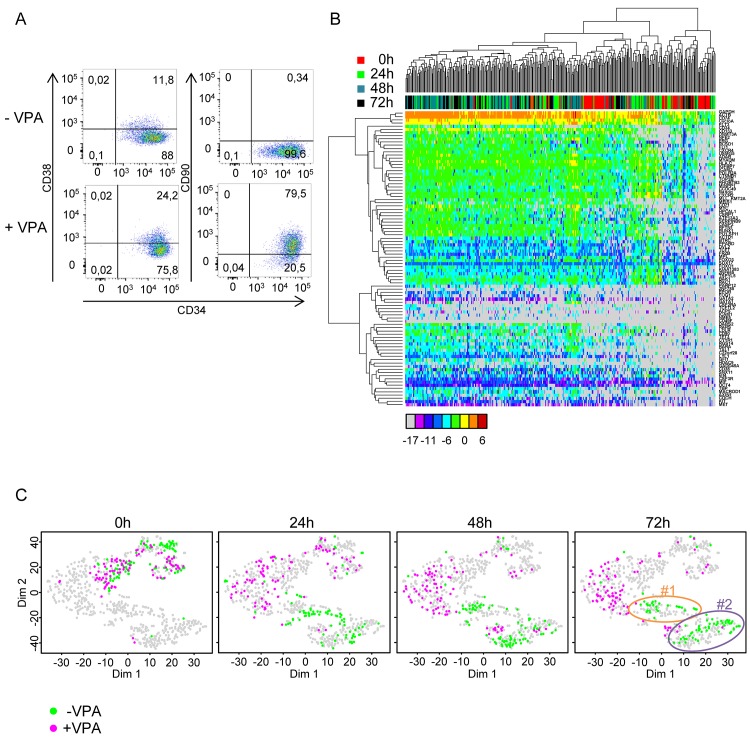
Individual cells take lineage commitment decisions in a way that is not necessarily uniform. We address this issue by characterising transcriptional changes in cord blood-derived CD34+ cells at the single-cell level and integrating data with cell division history and morphological changes determined by time-lapse microscopy. We show that major transcriptional changes leading to a multilineage-primed gene expression state occur very rapidly during the first cell cycle. One of the 2 stable lineage-primed patterns emerges gradually in each cell with variable timing. Some cells reach a stable morphology and molecular phenotype by the end of the first cell cycle and transmit it clonally. Others fluctuate between the 2 phenotypes over several cell cycles. Our analysis highlights the dynamic nature and variable timing of cell fate commitment in hematopoietic cells, links the gene expression pattern to cell morphology, and identifies a new category of cells with fluctuating phenotypic characteristics, demonstrating the complexity of the fate decision process (which is different from a simple binary switch between 2 options, as it is usually envisioned).
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

References
-
- Kawamoto H, Katsura Y. A new paradigm for hematopoietic cell lineages: revision of the classical concept of the myeloid-lymphoid dichotomy. Trends in immunology. 2009;30(5):193–200. doi: 10.1016/j.it.2009.03.001 . - DOI - PubMed
-
- Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404(6774):193–7. doi: 10.1038/35004599 . - DOI - PubMed
-
- Iwasaki H, Somoza C, Shigematsu H, Duprez EA, Iwasaki-Arai J, Mizuno S, et al. Distinctive and indispensable roles of PU.1 in maintenance of hematopoietic stem cells and their differentiation. Blood. 2005;106(5):1590–600. doi: 10.1182/blood-2005-03-0860 ; PubMed Central PMCID: PMC1895212. - DOI - PMC - PubMed
-
- Kondo M, Weissman IL, Akashi K. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell. 1997;91(5):661–72. . - PubMed
-
- Nutt SL, Metcalf D, D'Amico A, Polli M, Wu L. Dynamic regulation of PU.1 expression in multipotent hematopoietic progenitors. The Journal of experimental medicine. 2005;201(2):221–31. doi: 10.1084/jem.20041535 ; PubMed Central PMCID: PMC2212785. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
