

ReMixT: clone-specific genomic structure estimation in cancer
- PMID: 28750660
- PMCID: PMC5530528
- DOI: 10.1186/s13059-017-1267-2
ReMixT: clone-specific genomic structure estimation in cancer
Erratum in
-
Correction to: ReMixT: clone-specific genomic structure estimation in cancer.Genome Biol. 2017 Oct 6;18(1):188. doi: 10.1186/s13059-017-1327-7. Genome Biol. 2017. PMID: 28985744 Free PMC article. No abstract available.
Abstract
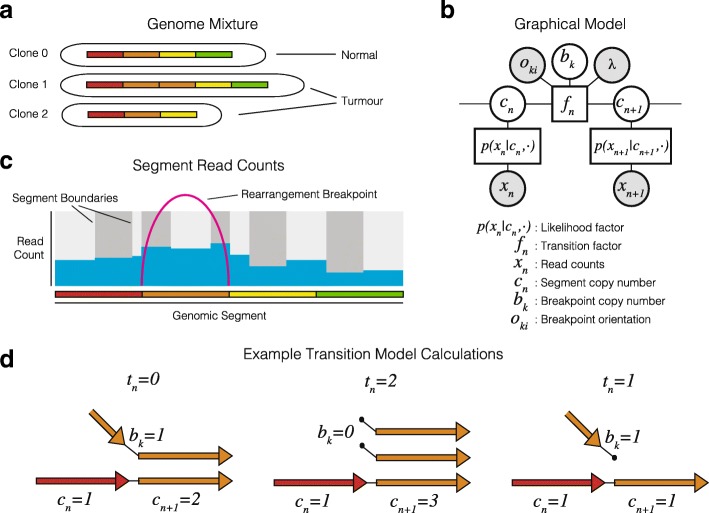
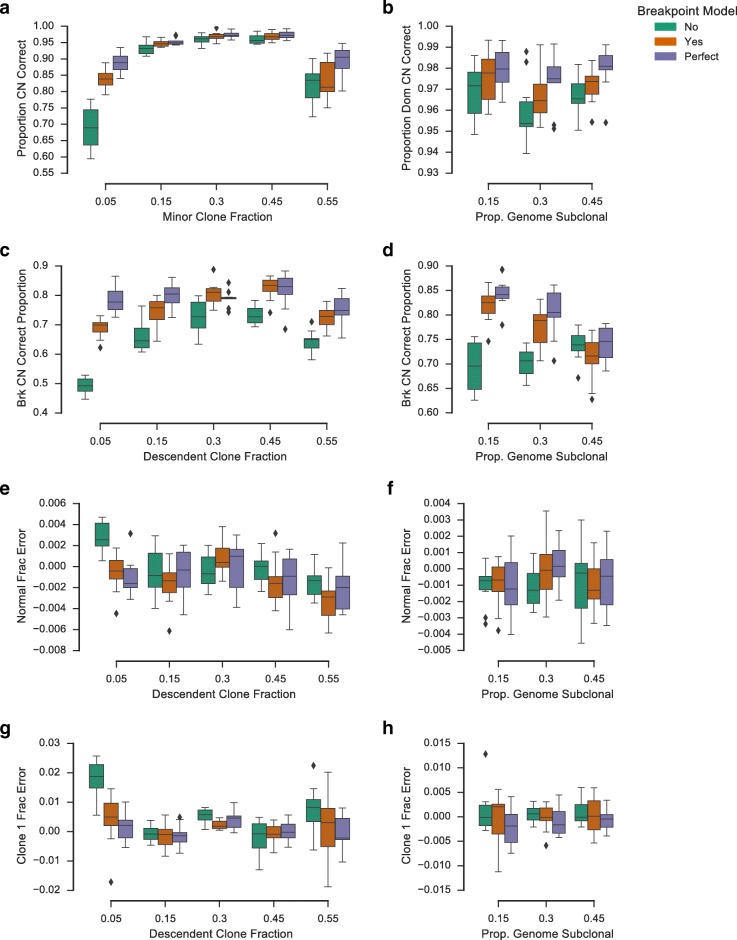
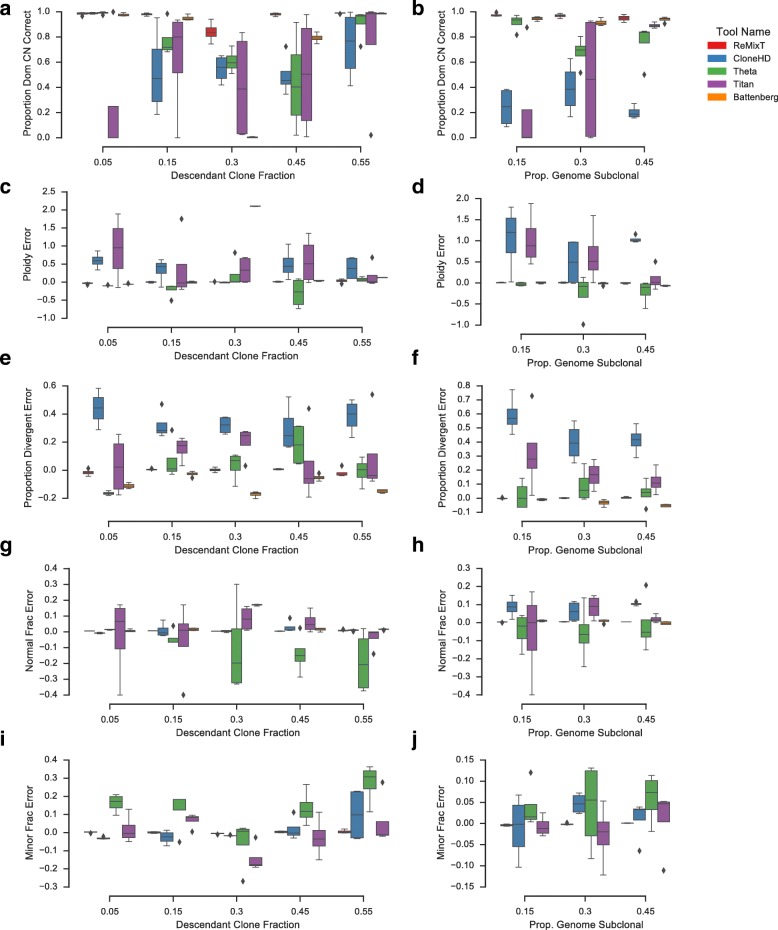
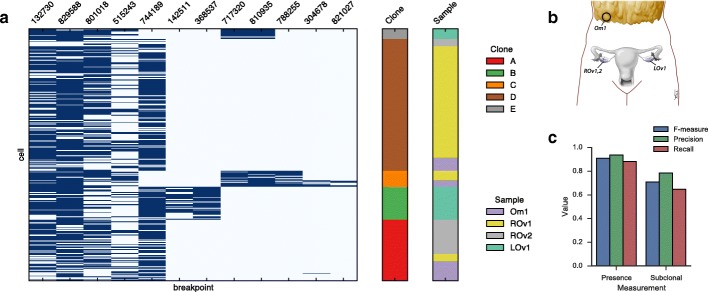
Somatic evolution of malignant cells produces tumors composed of multiple clonal populations, distinguished in part by rearrangements and copy number changes affecting chromosomal segments. Whole genome sequencing mixes the signals of sampled populations, diluting the signals of clone-specific aberrations, and complicating estimation of clone-specific genotypes. We introduce ReMixT, a method to unmix tumor and contaminating normal signals and jointly predict mixture proportions, clone-specific segment copy number, and clone specificity of breakpoints. ReMixT is free, open-source software and is available at http://bitbucket.org/dranew/remixt .
Keywords: Cancer genomics; Copy number variation; DNA sequencing; Genomic rearrangement; Tumour heterogeneity.
Conflict of interest statement
Ethics approval and consent to participate
Ethical approval was obtained from the University of British Columbia (UBC) Research Ethics Board (H08-01411 NGS Huntsman). Women undergoing debulking surgery (primary or recurrent) for carcinoma of ovarian, peritoneal, and/or fallopian tube origin were approached for informed consent for the banking of tumour tissue. All experimental methods comply with the Helsinki Declaration.
Anonymized tumour tissue from women aged 26–82 undergoing surgery or diagnostic core biopsy was collected with informed consent, according to procedures approved by the UBC Research Ethics Board (H06-00289 Breast Tumour Tissue Repository and H13-01125 Breast Xenograft Aparicio). All experimental methods comply with the Helsinki Declaration.
Female NOD/SCID interleukin-2 receptor gamma null (NSG) and NOD Rag-1 null interleukin-2 receptor gamma null (NRG) mice were bred and housed at the Animal Resource Centre at the British Columbia Cancer Research Centre and the Biological Resource Unit at the Cancer Research UK Cambridge Research Institute. Surgery was carried out on mice between the ages of 5–10 weeks. All experimental procedures were approved by the University of British Columbia Animal Care Committee and the University of Cambridge Animal Welfare and Ethical Review Committee.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- McPherson A, Shah SP, Sahinalp SC. deStruct: accurate rearrangement detection using breakpoint specific realignment. bioRxiv. 2017. https://doi.org/10.1101/117523. - DOI
-
- Ha G, Roth A, Khattra J, Ho J, Yap D, Prentice LM, Melnyk N, McPherson A, Bashashati A, Laks E, Biele J, Ding J, Le A, Rosner J, Shumansky K, Marra MA, Gilks CB, Huntsman DG, McAlpine JN, Aparicio S, Shah SP. TITAN: inference of copy number architectures in clonal cell populations from tumor whole-genome sequence data. Genome Res. 2014;24(11):1881–93. doi: 10.1101/gr.180281.114. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
