

Association of FGFR1 with ERα Maintains Ligand-Independent ER Transcription and Mediates Resistance to Estrogen Deprivation in ER+ Breast Cancer
- PMID: 28751448
- PMCID: PMC6681458
- DOI: 10.1158/1078-0432.CCR-17-1232
Association of FGFR1 with ERα Maintains Ligand-Independent ER Transcription and Mediates Resistance to Estrogen Deprivation in ER+ Breast Cancer
Erratum in
-
Correction: Association of FGFR1 with ERα Maintains Ligand-Independent ER Transcription and Mediates Resistance to Estrogen Deprivation in ER+ Breast Cancer.Clin Cancer Res. 2019 Feb 15;25(4):1433. doi: 10.1158/1078-0432.CCR-18-4268. Clin Cancer Res. 2019. PMID: 30770492 No abstract available.
Abstract
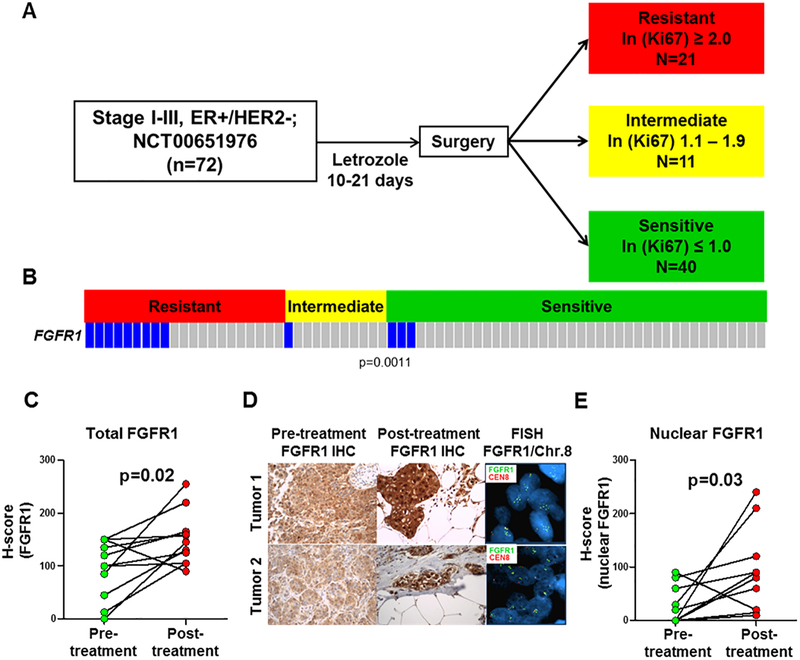
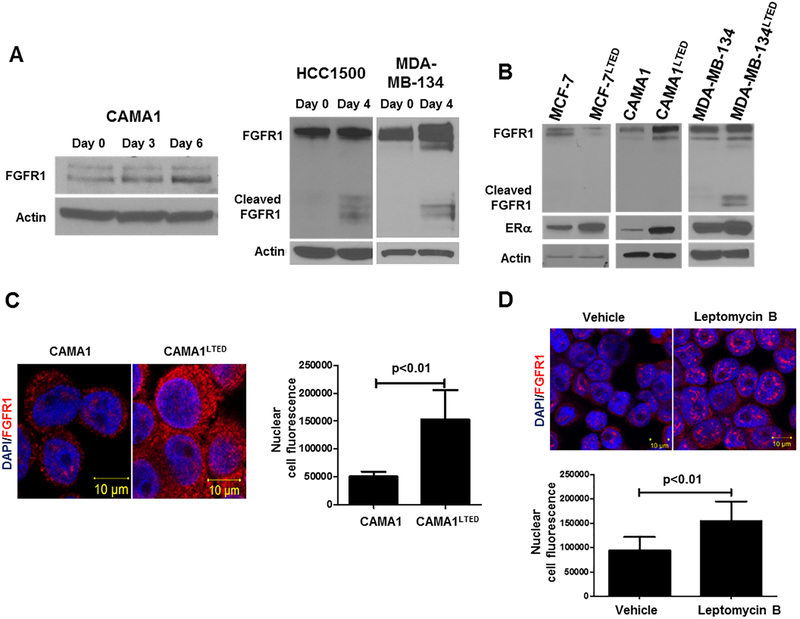
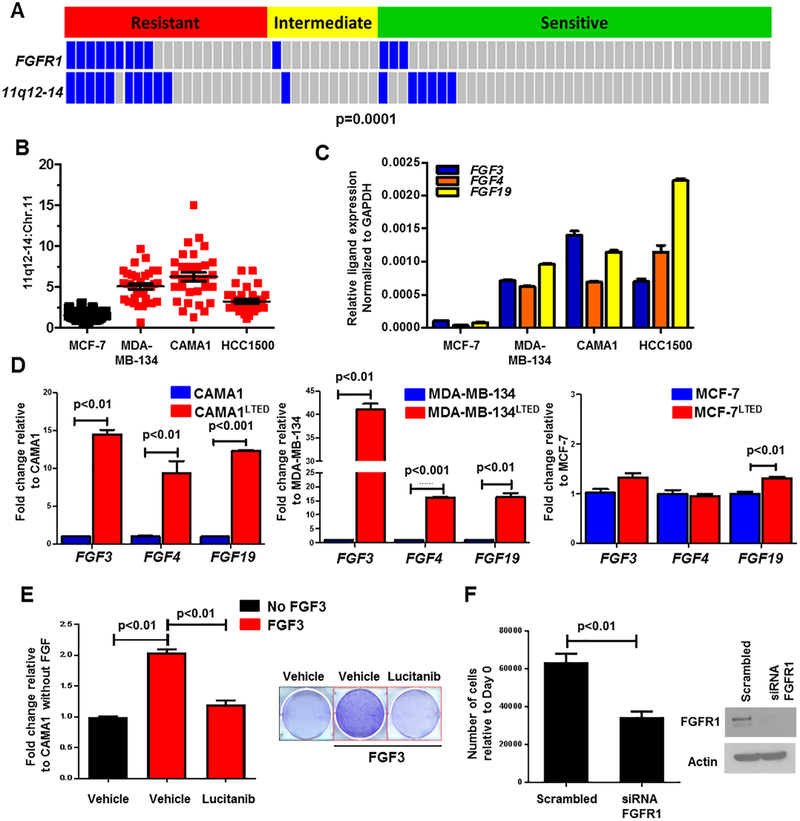
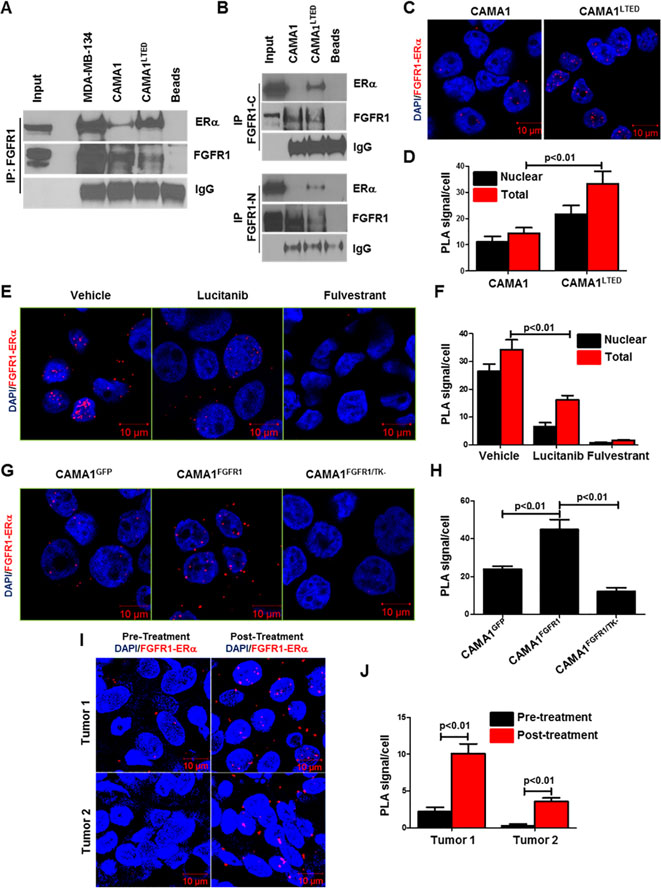
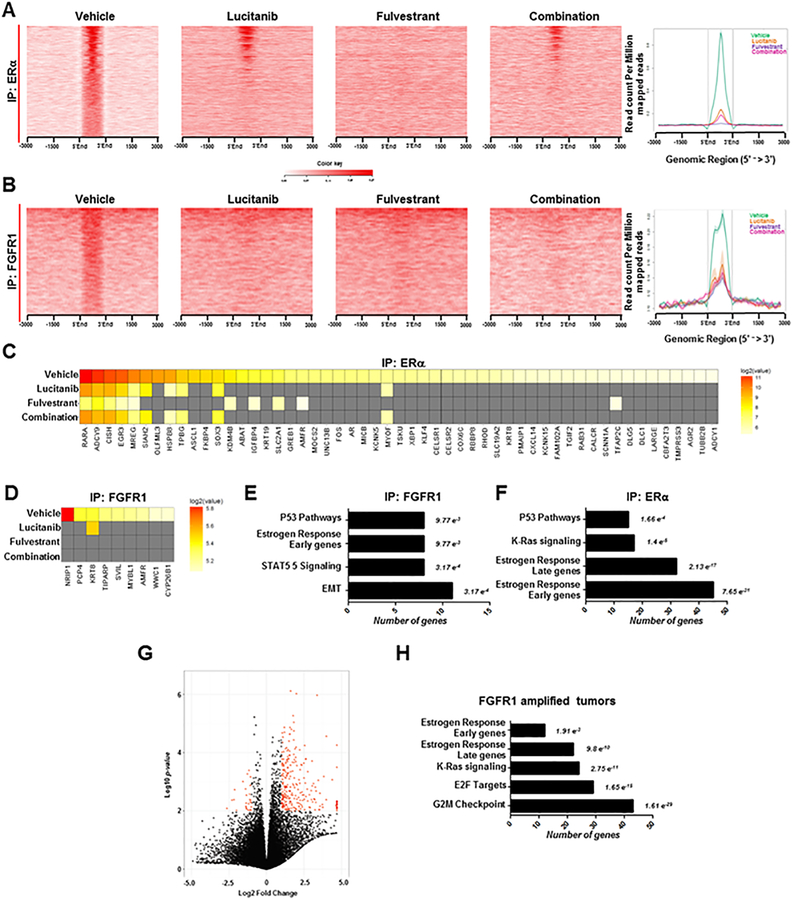
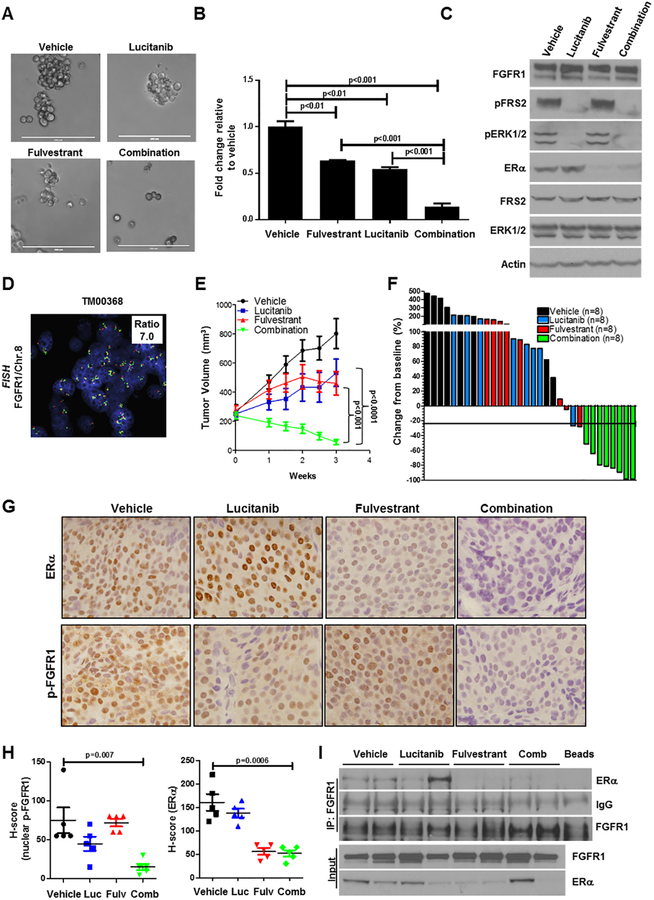
Purpose:FGFR1 amplification occurs in approximately 15% of estrogen receptor-positive (ER+) human breast cancers. We investigated mechanisms by which FGFR1 amplification confers antiestrogen resistance to ER+ breast cancer.Experimental Design: ER+ tumors from patients treated with letrozole before surgery were subjected to Ki67 IHC, FGFR1 FISH, and RNA sequencing (RNA-seq). ER+/FGFR1-amplified breast cancer cells, and patient-derived xenografts (PDX) were treated with FGFR1 siRNA or the FGFR tyrosine kinase inhibitor lucitanib. Endpoints were cell/xenograft growth, FGFR1/ERα association by coimmunoprecipitation and proximity ligation, ER genomic activity by ChIP sequencing, and gene expression by RT-PCR.Results: ER+/FGFR1-amplified tumors in patients treated with letrozole maintained cell proliferation (Ki67). Estrogen deprivation increased total and nuclear FGFR1 and FGF ligands expression in ER+/FGFR1-amplified primary tumors and breast cancer cells. In estrogen-free conditions, FGFR1 associated with ERα in tumor cell nuclei and regulated the transcription of ER-dependent genes. This association was inhibited by a kinase-dead FGFR1 mutant and by treatment with lucitanib. ChIP-seq analysis of estrogen-deprived ER+/FGFR1-amplified cells showed binding of FGFR1 and ERα to DNA. Treatment with fulvestrant and/or lucitanib reduced FGFR1 and ERα binding to DNA. RNA-seq data from FGFR1-amplified patients' tumors treated with letrozole showed enrichment of estrogen response and E2F target genes. Finally, growth of ER+/FGFR1-amplified cells and PDXs was more potently inhibited by fulvestrant and lucitanib combined than each drug alone.Conclusions: These data suggest the ERα pathway remains active in estrogen-deprived ER+/FGFR1-amplified breast cancers. Therefore, these tumors are endocrine resistant and should be candidates for treatment with combinations of ER and FGFR antagonists. Clin Cancer Res; 23(20); 6138-50. ©2017 AACR.
©2017 American Association for Cancer Research.
Conflict of interest statement
Figures
References
-
- Courjal F, Cuny M, Simony-Lafontaine J, Louason G, Speiser P, Zeillinger R, et al. Mapping of DNA amplifications at 15 chromosomal localizations in 1875 breast tumors: definition of phenotypic groups. Cancer Res. 1997;57(19):4360–7. - PubMed
-
- Jacquemier J, Adelaide J, Parc P, Penault-Llorca F, Planche J, deLapeyriere O, et al. Expression of the FGFR1 gene in human breast-carcinoma cells. Int J Cancer. 1994;59(3):373–8. - PubMed
-
- Reis-Filho JS, Simpson PT, Turner NC, Lambros MB, Jones C, Mackay A, et al. FGFR1 emerges as a potential therapeutic target for lobular breast carcinomas. Clin Cancer Res. 2006;12(22):6652–62. - PubMed
-
- Gorringe KL, Jacobs S, Thompson ER, Sridhar A, Qiu W, Choong DY, et al. High-resolution single nucleotide polymorphism array analysis of epithelial ovarian cancer reveals numerous microdeletions and amplifications. Clin Cancer Res. 2007;13(16):4731–9. - PubMed
-
- Simon R, Richter J, Wagner U, Fijan A, Bruderer J, Schmid U, et al. High-throughput tissue microarray analysis of 3p25 (RAF1) and 8p12 (FGFR1) copy number alterations in urinary bladder cancer. Cancer Res. 2001;61(11):4514–9. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
