

Automated incorporation of pairwise dependency in transcription factor binding site prediction using dinucleotide weight tensors
- PMID: 28753602
- PMCID: PMC5550003
- DOI: 10.1371/journal.pcbi.1005176
Automated incorporation of pairwise dependency in transcription factor binding site prediction using dinucleotide weight tensors
Abstract
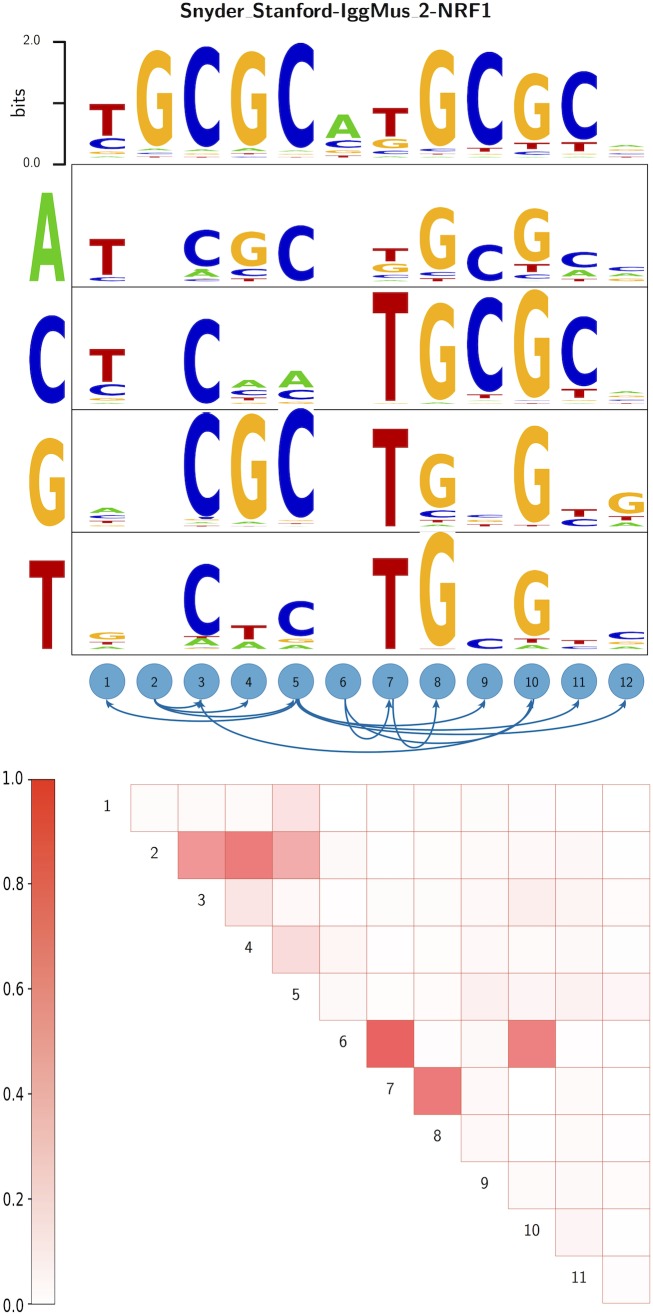
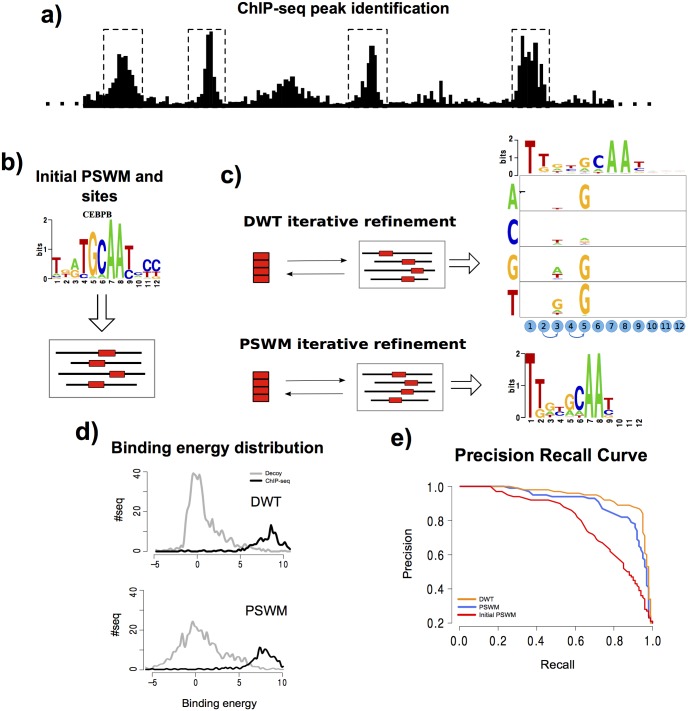
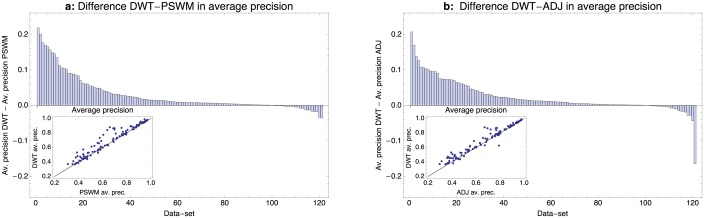
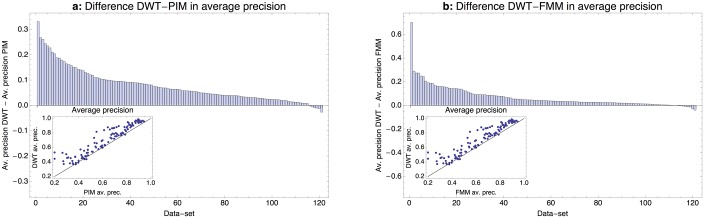
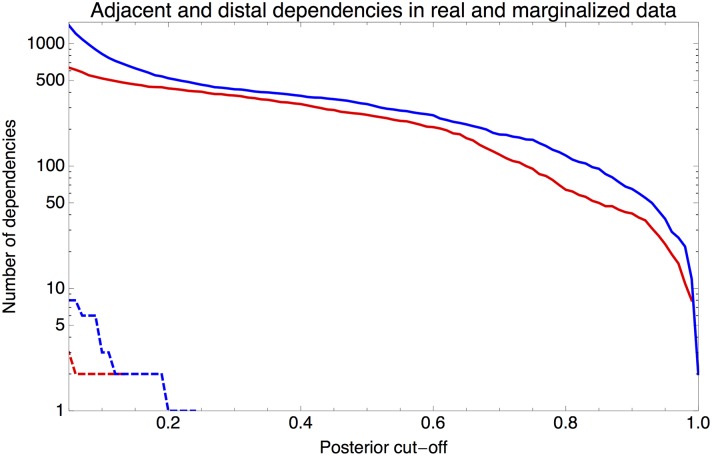
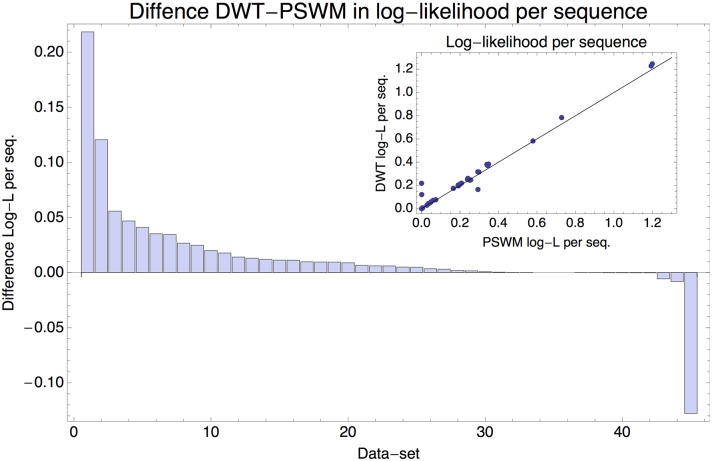
Gene regulatory networks are ultimately encoded by the sequence-specific binding of (TFs) to short DNA segments. Although it is customary to represent the binding specificity of a TF by a position-specific weight matrix (PSWM), which assumes each position within a site contributes independently to the overall binding affinity, evidence has been accumulating that there can be significant dependencies between positions. Unfortunately, methodological challenges have so far hindered the development of a practical and generally-accepted extension of the PSWM model. On the one hand, simple models that only consider dependencies between nearest-neighbor positions are easy to use in practice, but fail to account for the distal dependencies that are observed in the data. On the other hand, models that allow for arbitrary dependencies are prone to overfitting, requiring regularization schemes that are difficult to use in practice for non-experts. Here we present a new regulatory motif model, called dinucleotide weight tensor (DWT), that incorporates arbitrary pairwise dependencies between positions in binding sites, rigorously from first principles, and free from tunable parameters. We demonstrate the power of the method on a large set of ChIP-seq data-sets, showing that DWTs outperform both PSWMs and motif models that only incorporate nearest-neighbor dependencies. We also demonstrate that DWTs outperform two previously proposed methods. Finally, we show that DWTs inferred from ChIP-seq data also outperform PSWMs on HT-SELEX data for the same TF, suggesting that DWTs capture inherent biophysical properties of the interactions between the DNA binding domains of TFs and their binding sites. We make a suite of DWT tools available at dwt.unibas.ch, that allow users to automatically perform 'motif finding', i.e. the inference of DWT motifs from a set of sequences, binding site prediction with DWTs, and visualization of DWT 'dilogo' motifs.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
