

Peripheral complement interactions with amyloid β peptide in Alzheimer's disease: 2. Relationship to amyloid β immunotherapy
- PMID: 28755839
- PMCID: PMC5881571
- DOI: 10.1016/j.jalz.2017.04.015
Peripheral complement interactions with amyloid β peptide in Alzheimer's disease: 2. Relationship to amyloid β immunotherapy
Abstract
Introduction: Our previous studies have shown that amyloid β peptide (Aβ) is subject to complement-mediated clearance from the peripheral circulation, and that this mechanism is deficient in Alzheimer's disease. The mechanism should be enhanced by Aβ antibodies that form immune complexes (ICs) with Aβ, and therefore may be relevant to current Aβ immunotherapy approaches.
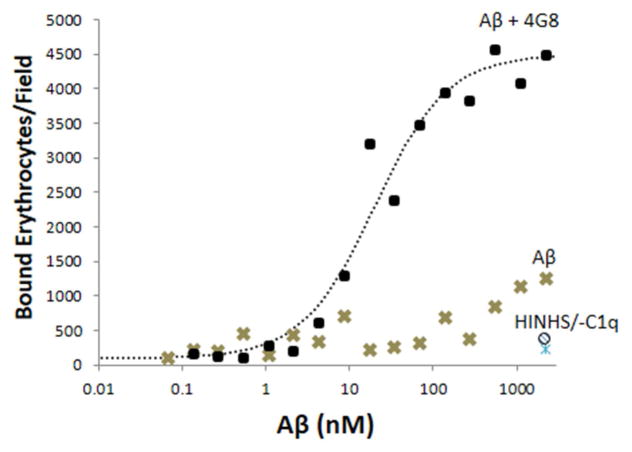
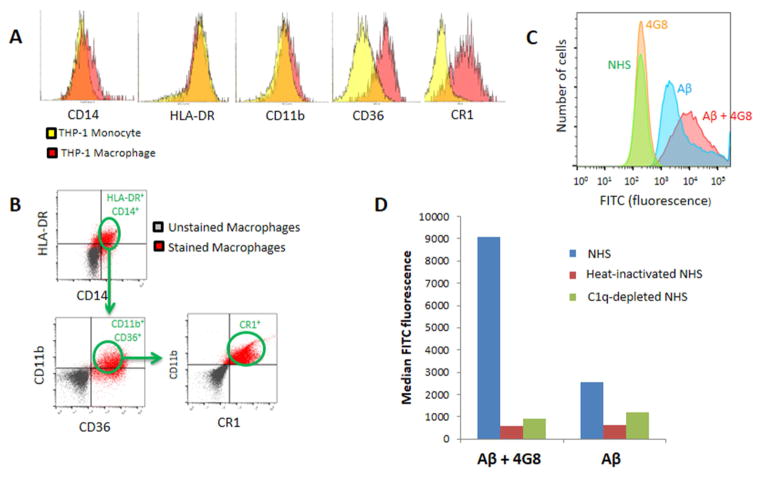
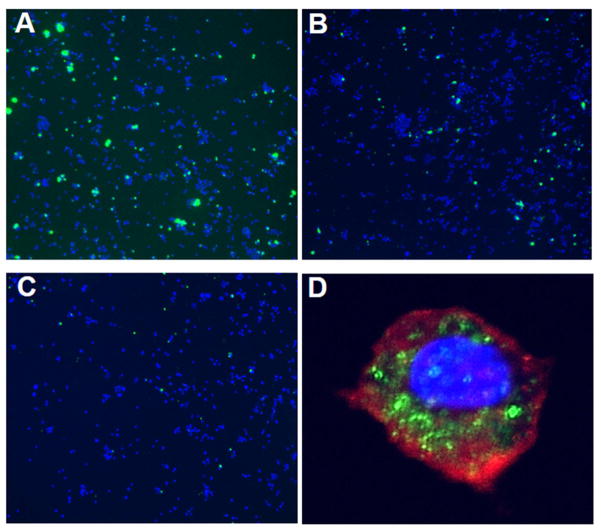
Methods: Multidisciplinary methods were employed to demonstrate enhanced complement-mediated capture of Aβ antibody immune complexes compared with Aβ alone in both erythrocytes and THP1-derived macrophages.
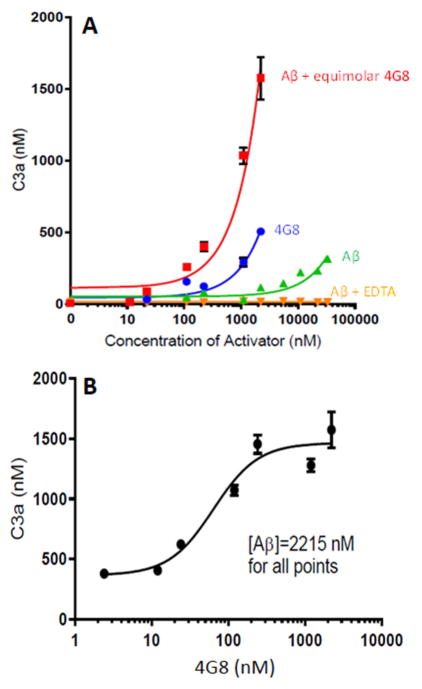
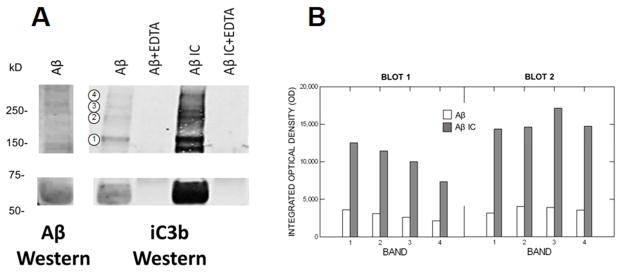
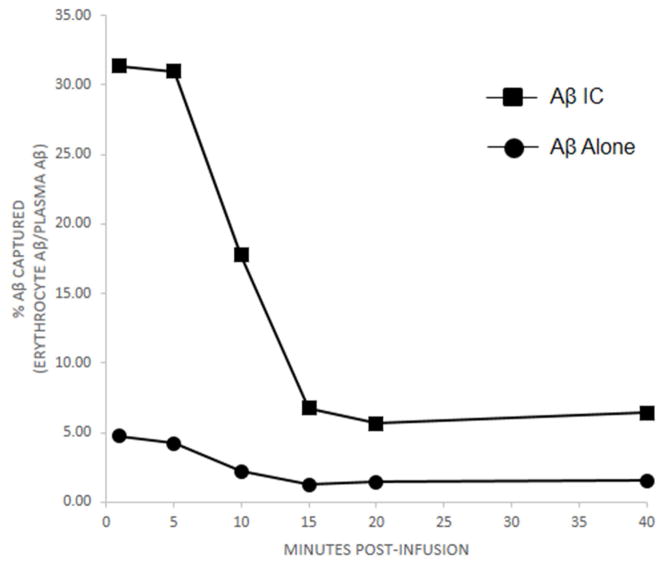
Results: Aβ antibodies dramatically increased complement activation and opsonization of Aβ, followed by commensurately enhanced Aβ capture by human erythrocytes and macrophages. These in vitro findings were consistent with enhanced peripheral clearance of intravenously administered Aβ antibody immune complexes in nonhuman primates.
Discussion: Together with our previous results, showing significant Alzheimer's disease deficits in peripheral Aβ clearance, the present findings strongly suggest that peripheral mechanisms should not be ignored as contributors to the effects of Aβ immunotherapy.
Keywords: Alzheimer's disease; Amyloid β peptide; Aβ immunotherapy; Blood; Complement; Complement receptor 1; Erythrocyte; Human; Macrophage.
Copyright © 2017 the Alzheimer's Association. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures
Similar articles
-
Peripheral complement interactions with amyloid β peptide: Erythrocyte clearance mechanisms.Alzheimers Dement. 2017 Dec;13(12):1397-1409. doi: 10.1016/j.jalz.2017.03.010. Epub 2017 May 2. Alzheimers Dement. 2017. PMID: 28475854 Free PMC article.
-
A novel role for SHARPIN in amyloid-β phagocytosis and inflammation by peripheral blood-derived macrophages in Alzheimer's disease.Neurobiol Aging. 2020 Sep;93:131-141. doi: 10.1016/j.neurobiolaging.2020.02.001. Epub 2020 Feb 13. Neurobiol Aging. 2020. PMID: 32165044
-
Targeting beta-amyloid pathology in Alzheimer's disease with Abeta immunotherapy.Neurotherapeutics. 2008 Jul;5(3):415-20. doi: 10.1016/j.nurt.2008.05.013. Neurotherapeutics. 2008. PMID: 18625453 Free PMC article. Review.
-
Methods to monitor monocytes-mediated amyloid-beta uptake and phagocytosis in the context of adjuvanted immunotherapies.J Immunol Methods. 2015 Sep;424:64-79. doi: 10.1016/j.jim.2015.05.002. Epub 2015 May 19. J Immunol Methods. 2015. PMID: 26002154
-
'Clinical trials in Alzheimer's disease': immunotherapy approaches.J Neurochem. 2012 Jan;120 Suppl 1:186-193. doi: 10.1111/j.1471-4159.2011.07458.x. Epub 2011 Nov 28. J Neurochem. 2012. Retraction in: J Neurochem. 2021 Aug;158(3):821. doi: 10.1111/jnc.15435. PMID: 21883222 Retracted. Review.
Cited by
-
Systematic analysis of dark and camouflaged genes reveals disease-relevant genes hiding in plain sight.Genome Biol. 2019 May 20;20(1):97. doi: 10.1186/s13059-019-1707-2. Genome Biol. 2019. PMID: 31104630 Free PMC article.
-
Clearance of amyloid-beta with bispecific antibody constructs bound to erythrocytes.Alzheimers Dement (N Y). 2020 Aug 27;6(1):e12067. doi: 10.1002/trc2.12067. eCollection 2020. Alzheimers Dement (N Y). 2020. PMID: 32885023 Free PMC article.
-
Elovanoids counteract oligomeric β-amyloid-induced gene expression and protect photoreceptors.Proc Natl Acad Sci U S A. 2019 Nov 26;116(48):24317-24325. doi: 10.1073/pnas.1912959116. Epub 2019 Nov 11. Proc Natl Acad Sci U S A. 2019. PMID: 31712409 Free PMC article.
-
Complement-Mediated Events in Alzheimer's Disease: Mechanisms and Potential Therapeutic Targets.J Immunol. 2020 Jan 15;204(2):306-315. doi: 10.4049/jimmunol.1901068. J Immunol. 2020. PMID: 31907273 Free PMC article. Review.
-
The good, the bad, and the opportunities of the complement system in neurodegenerative disease.J Neuroinflammation. 2020 Nov 25;17(1):354. doi: 10.1186/s12974-020-02024-8. J Neuroinflammation. 2020. PMID: 33239010 Free PMC article. Review.
References
-
- Bard F, Cannon C, Barbour R, Burke RL, Games D, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000 Aug;6(8):916–9. - PubMed
-
- Gilman S, Koller M, Black RS, et al. Clinical effects of A-beta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64(9):1553–1562. - PubMed
-
- Boche D, Denham N, Holmes C, Nicoll JA. Neuropathology after active Abeta42 immunotherapy: implications for Alzheimer’s disease pathogenesis. Acta Neuropathol. 2010 Sep;120(3):369–84. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
