

Clinical profile of unclassifiable interstitial lung disease: Comparison with chronic fibrosing idiopathic interstitial pneumonias
- PMID: 28758849
- PMCID: PMC6011274
- DOI: 10.1177/0300060517719767
Clinical profile of unclassifiable interstitial lung disease: Comparison with chronic fibrosing idiopathic interstitial pneumonias
Abstract
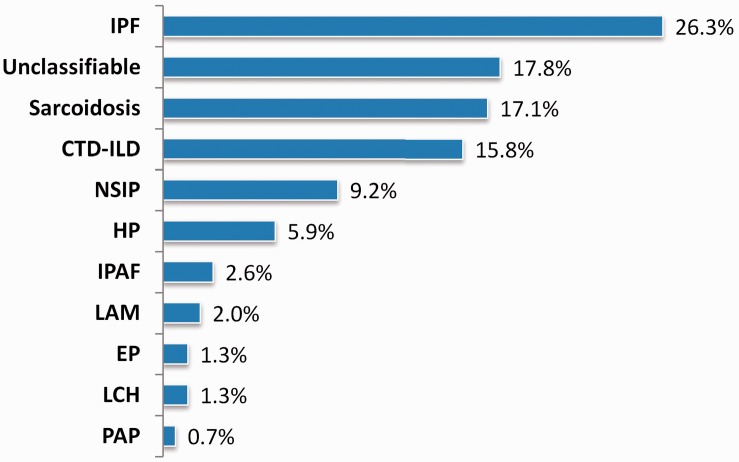
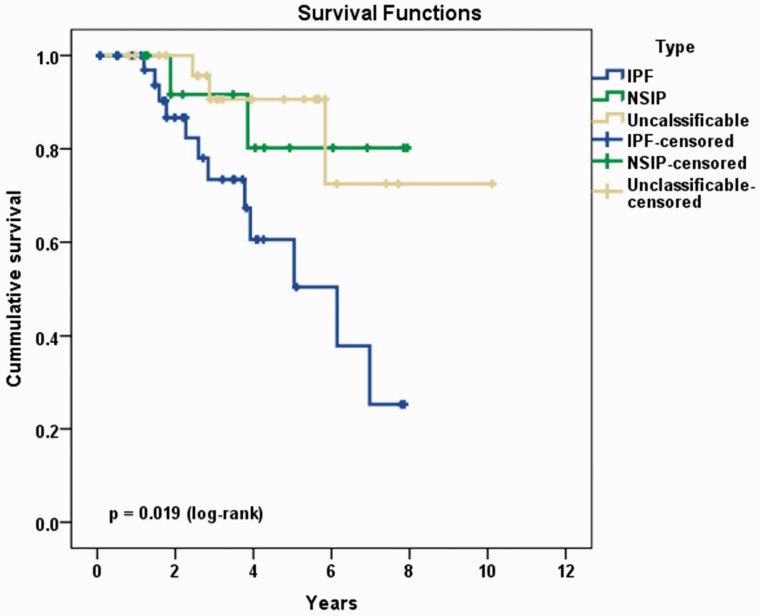
Objective Unclassifiable interstitial lung disease (ILD) is a common problem in clinical practice. These patients pose a distinct challenge with regard to appropriate evaluation and management. We investigated the clinical features and prognosis of unclassifiable ILD and compared its clinical profile with that of idiopathic pulmonary fibrosis (IPF) and idiopathic nonspecific interstitial pneumonia (NSIP). Methods Patients with IPF (n = 40), NSIP (n = 14), and unclassifiable ILD (n = 27) were selected from an ongoing database. Baseline clinical features, pulmonary function, and the extent of fibrosis on high-resolution computed tomography (HRCT) were evaluated. Mortality was estimated based on the ILD-Gender, Age, Physiology (ILD-GAP) index and composite physiologic index (CPI). Results IPF was associated with the worst survival (hazard ratio [HR] = 4.361 compared with NSIP), followed by unclassifiable cases (HR = 1.251 compared with NSIP). Increasing mortality was significantly impacted by age (HR = 1.04 per 1-year increase), lower carbon monoxide diffusing capacity of the lung (HR = 0.97), HRCT interstitial score (HR = 1.119 per 1-point increase), ILD-GAP score (HR = 1.570 per 1-point increase), and CPI (HR = 1.039 per 1-point increase). Conclusions Patients with unclassifiable ILD had an intermediate prognosis between that of IPF and NSIP. Patients at high risk of mortality can be identified using baseline clinical, physiological, and radiological features.
Keywords: Idiopathic interstitial pneumonia; high-resolution computed tomography; idiopathic nonspecific interstitial pneumonia; idiopathic pulmonary fibrosis; mortality; unclassifiable interstitial lung disease.
Figures
Similar articles
-
Comparison of clinical courses and mortality of connective tissue disease-associated interstitial pneumonias and chronic fibrosing idiopathic interstitial pneumonias.Kaohsiung J Med Sci. 2019 Jun;35(6):365-372. doi: 10.1002/kjm2.12066. Epub 2019 Mar 26. Kaohsiung J Med Sci. 2019. PMID: 30913371 Free PMC article.
-
A cohort study of Danish patients with interstitial lung diseases: burden, severity, treatment and survival.Dan Med J. 2015 Apr;62(4):B5069. Dan Med J. 2015. PMID: 25872544 Review.
-
Unclassifiable interstitial lung diseases: Clinical characteristics and survival.Respirology. 2017 Apr;22(3):494-500. doi: 10.1111/resp.12931. Epub 2016 Nov 6. Respirology. 2017. PMID: 28297158
-
High resolution computed tomography pattern of usual interstitial pneumonia in rheumatoid arthritis-associated interstitial lung disease: Relationship to survival.Respir Med. 2017 May;126:100-104. doi: 10.1016/j.rmed.2017.03.027. Epub 2017 Mar 30. Respir Med. 2017. PMID: 28427540
-
Challenges in the classification of fibrotic ILD.Sarcoidosis Vasc Diffuse Lung Dis. 2015 Aug 3;32 Suppl 1:4-9. Sarcoidosis Vasc Diffuse Lung Dis. 2015. PMID: 26237436 Review.
Cited by
-
Observational findings of transbronchial lung biopsy in patients with interstitial lung disease: a retrospective study in Aleppo University Hospital.Ann Med Surg (Lond). 2023 Feb 17;85(2):146-152. doi: 10.1097/MS9.0000000000000180. eCollection 2023 Feb. Ann Med Surg (Lond). 2023. PMID: 36845790 Free PMC article.
-
miR-21 Exerts Anti-proliferative and Pro-apoptotic Effects in LPS-induced WI-38 Cells via Directly Targeting TIMP3.Cell Biochem Biophys. 2021 Dec;79(4):781-790. doi: 10.1007/s12013-021-00987-w. Epub 2021 May 3. Cell Biochem Biophys. 2021. PMID: 33942238
-
Efficacy of Pirfenidone vs. Placebo in Unclassifiable Interstitial Lung Disease, by Surgical Lung Biopsy Status: Data From a post-hoc Analysis.Front Med (Lausanne). 2022 Jun 17;9:897102. doi: 10.3389/fmed.2022.897102. eCollection 2022. Front Med (Lausanne). 2022. PMID: 35783648 Free PMC article.
-
Management of Chronic Respiratory Failure in Interstitial Lung Diseases: Overview and Clinical Insights.Int J Med Sci. 2019 Jun 10;16(7):967-980. doi: 10.7150/ijms.32752. eCollection 2019. Int J Med Sci. 2019. PMID: 31341410 Free PMC article. Review.
-
Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases.Eur Respir Rev. 2018 Dec 21;27(150):180076. doi: 10.1183/16000617.0076-2018. Print 2018 Dec 31. Eur Respir Rev. 2018. PMID: 30578335 Free PMC article. Review.
References
-
- Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013; 188: 733–748. doi: 10.1164/rccm.201308-1483ST PMID: 24032382. - PMC - PubMed
-
- Raghu G, Weycker D, Edelsberg J, et al. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2006; 174: 810–816. - PubMed
-
- Ley B, Collard HR, King TE., Jr Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 183: 431–440. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
