

Tricetin Induces Apoptosis of Human Leukemic HL-60 Cells through a Reactive Oxygen Species-Mediated c-Jun N-Terminal Kinase Activation Pathway
- PMID: 28758971
- PMCID: PMC5578057
- DOI: 10.3390/ijms18081667
Tricetin Induces Apoptosis of Human Leukemic HL-60 Cells through a Reactive Oxygen Species-Mediated c-Jun N-Terminal Kinase Activation Pathway
Abstract
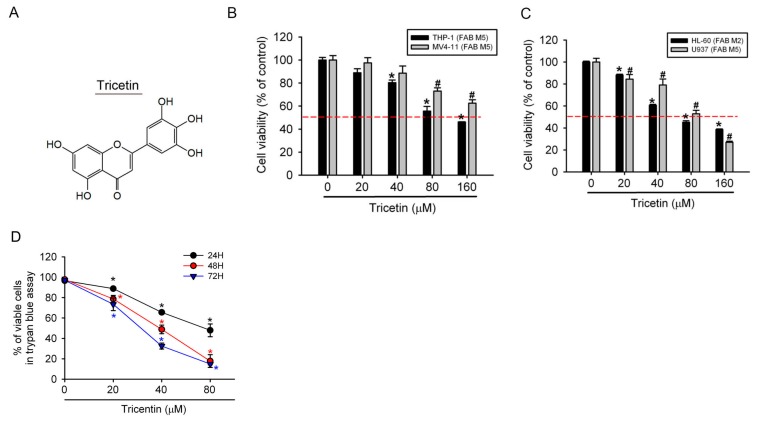
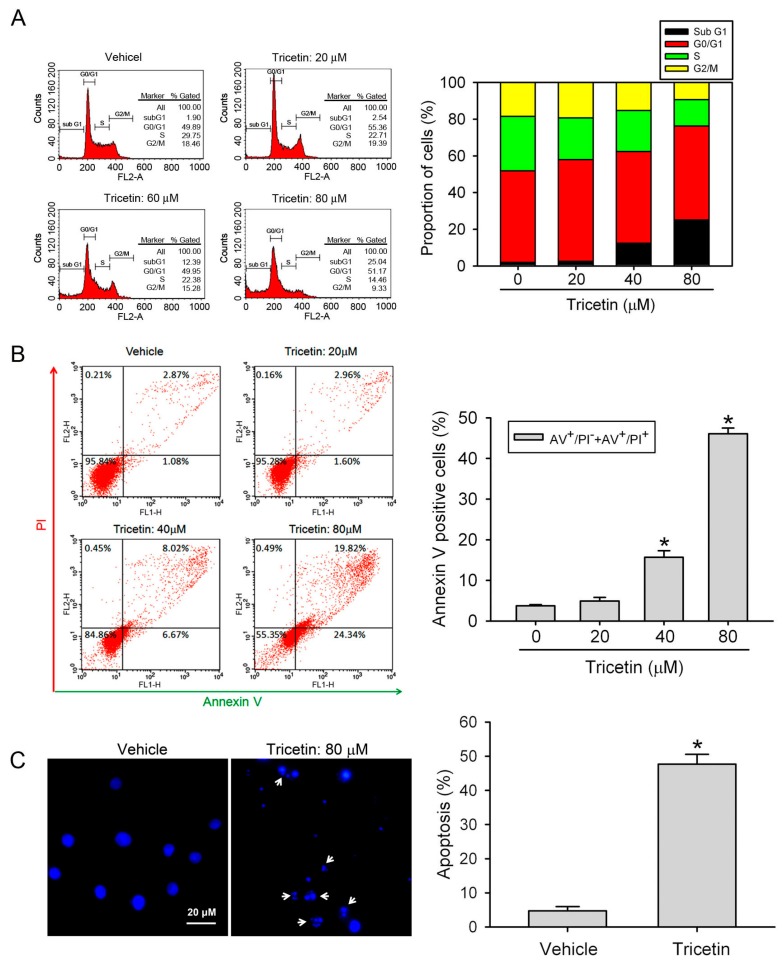
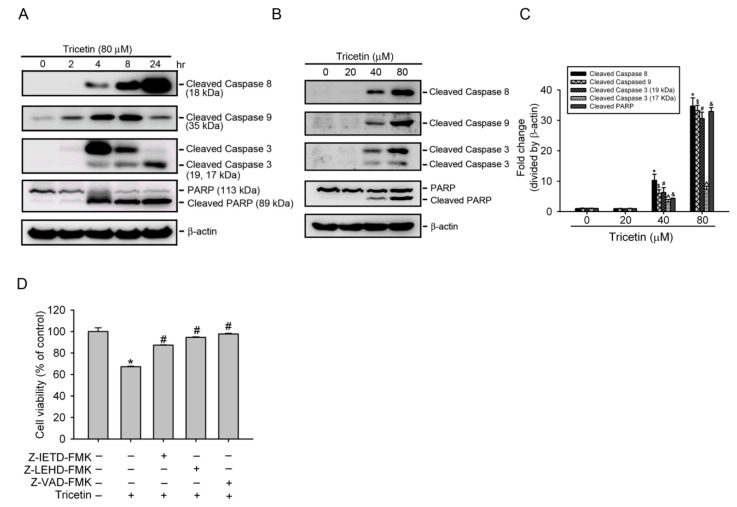
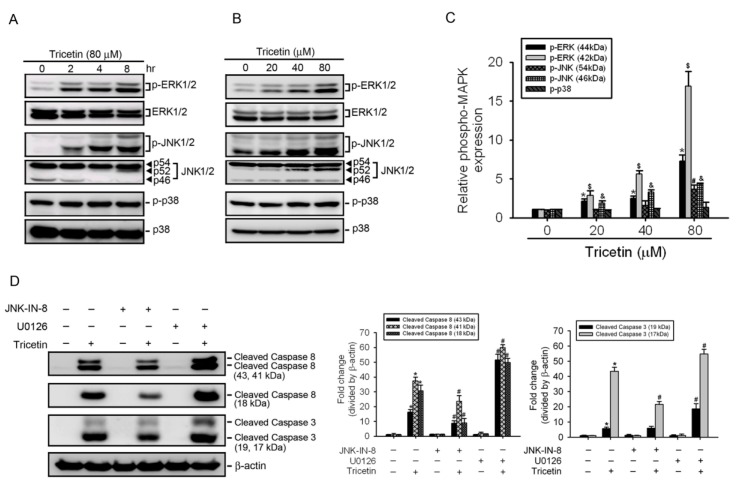
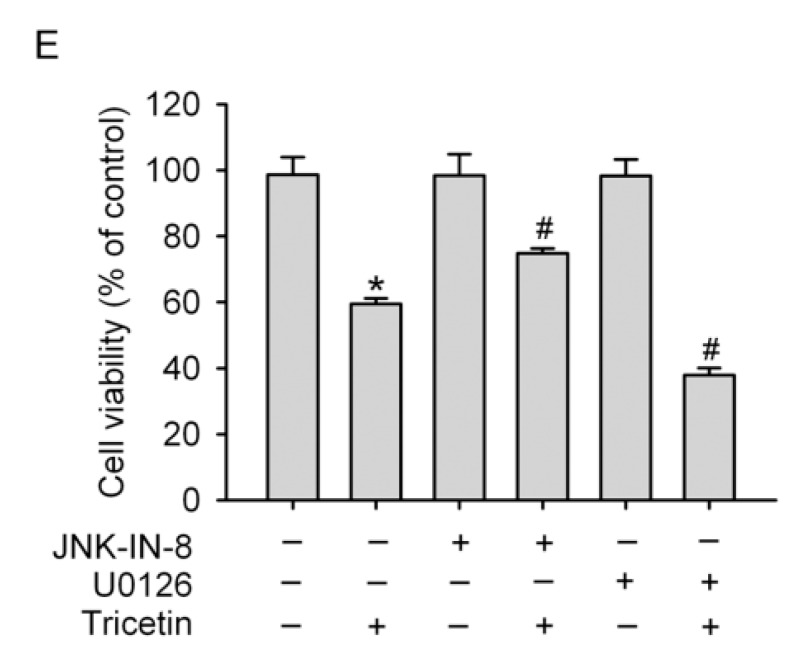
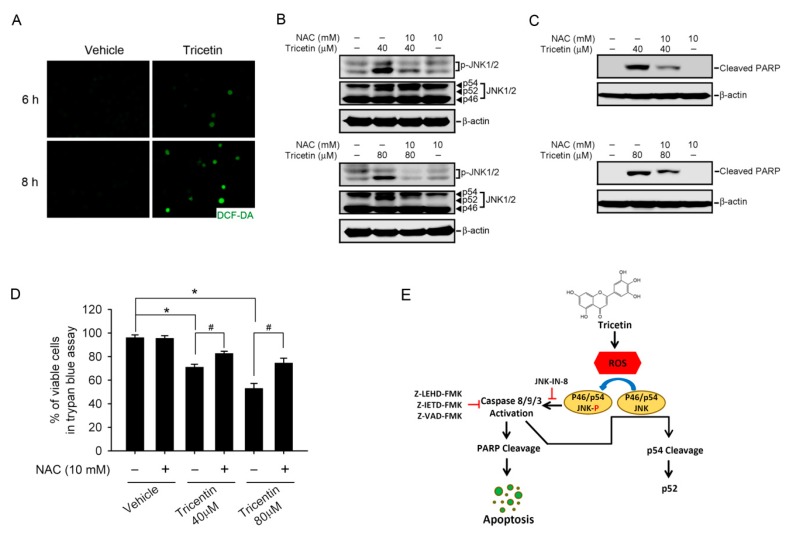
Tricetin is a dietary flavonoid with cytostatic properties and antimetastatic activities in various solid tumors. The anticancer effect of tricetin in nonsolid tumors remains unclear. Herein, the molecular mechanisms by which tricetin exerts its anticancer effects on acute myeloid leukemia (AML) cells were investigated. Results showed that tricetin inhibited cell viability in various types of AML cell lines. Tricetin induced morphological features of apoptosis such as chromatin condensation and phosphatidylserine (PS) externalization, and significantly activated proapoptotic signaling including caspase-8, -9, and -3 activation and poly(ADP-ribose) polymerase (PARP) cleavage in HL-60 AML cells. Of note, tricetin-induced cell growth inhibition was dramatically reversed by a pan caspase and caspase-8- and -9-specific inhibitors, suggesting that this compound mainly acts through a caspase-dependent pathway. Moreover, treatment of HL-60 cells with tricetin induced sustained activation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK), and inhibition of ERK and JNK by their specific inhibitors respectively promoted and abolished tricetin-induced cell apoptosis. Dichlorofluorescein (DCF) staining showed that intracellular reactive oxygen species (ROS) levels were higher in tricetin-treated HL-60 cells compared to the control group. Moreover, an ROS scavenger, N-acetylcysteine (NAC), reversed tricetin-induced JNK activation and subsequent cell apoptosis. In conclusion, our results indicated that tricetin induced cell death of leukemic HL-60 cells through induction of intracellular oxidative stress following activation of a JNK-mediated apoptosis pathway. A combination of tricetin and an ERK inhibitor may be a better strategy to enhance the anticancer activities of tricetin in AML.
Keywords: acute myeloid leukemia; apoptosis; c-Jun N-terminal kinase; reactive oxygen species; tricetin.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Burnett A.K., Hills R.K., Milligan D.W., Goldstone A.H., Prentice A.G., McMullin M.F., Duncombe A., Gibson B., Wheatley K. Attempts to optimize induction and consolidation treatment in acute myeloid leukemia: Results of the MRC AML12 trial. J. Clin. Oncol. 2010;28:586–595. doi: 10.1200/JCO.2009.22.9088. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
