

Mutation-induced loss of APP function causes GABAergic depletion in recessive familial Alzheimer's disease: analysis of Osaka mutation-knockin mice
- PMID: 28760161
- PMCID: PMC5537936
- DOI: 10.1186/s40478-017-0461-5
Mutation-induced loss of APP function causes GABAergic depletion in recessive familial Alzheimer's disease: analysis of Osaka mutation-knockin mice
Abstract
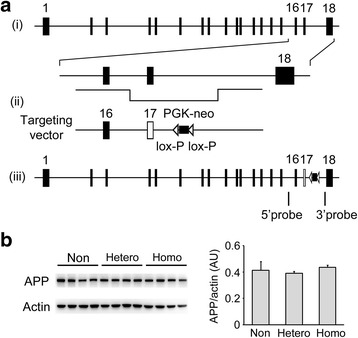
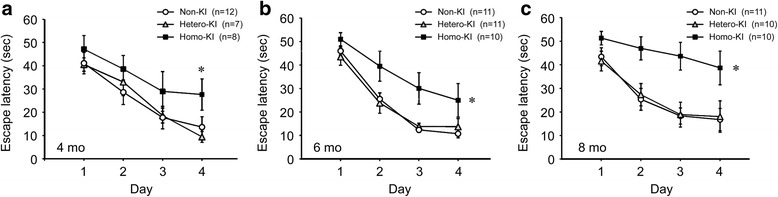
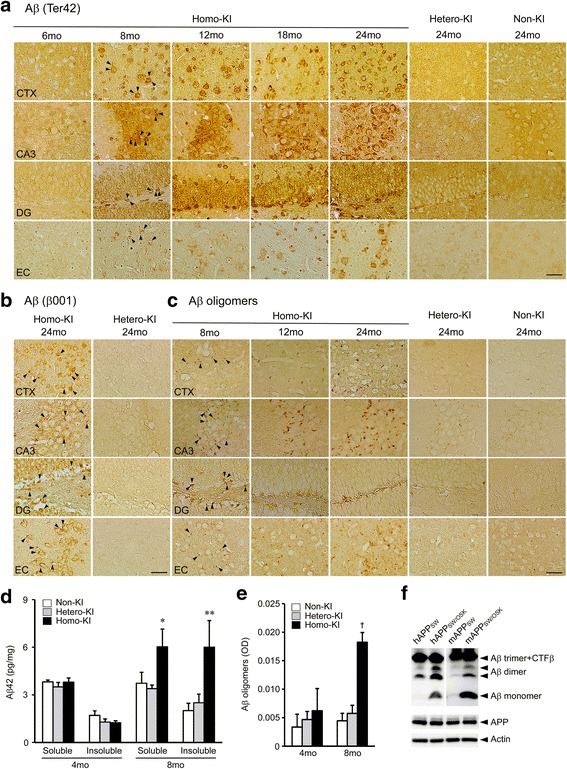
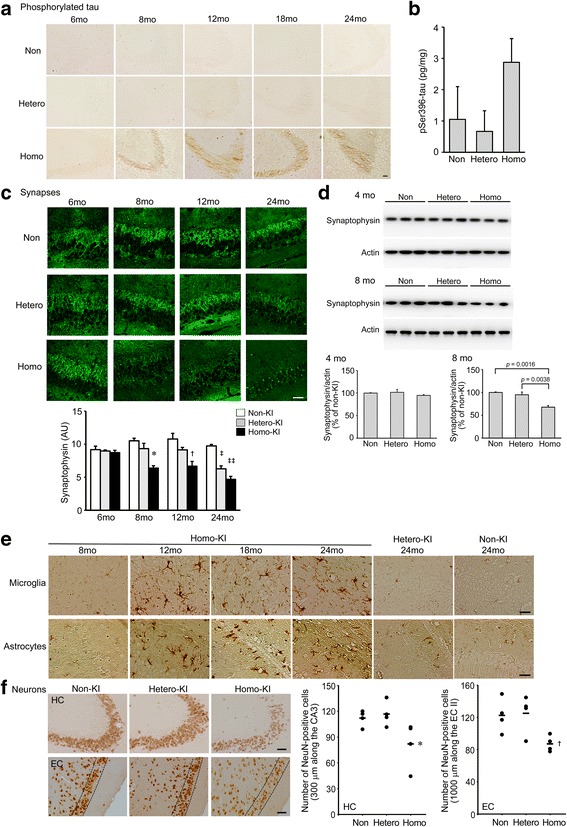
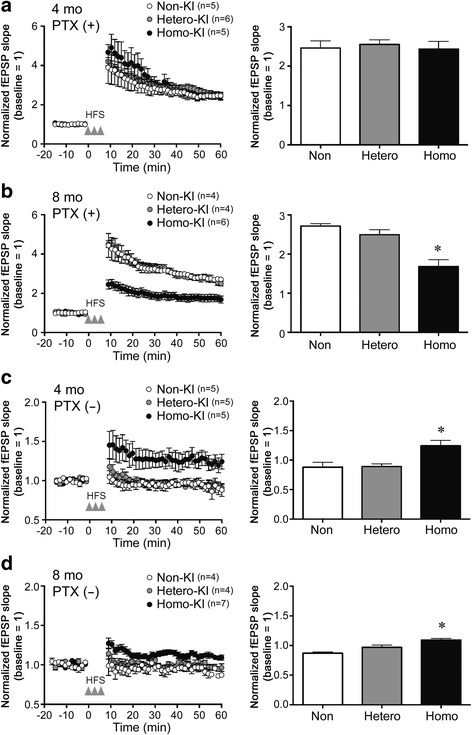
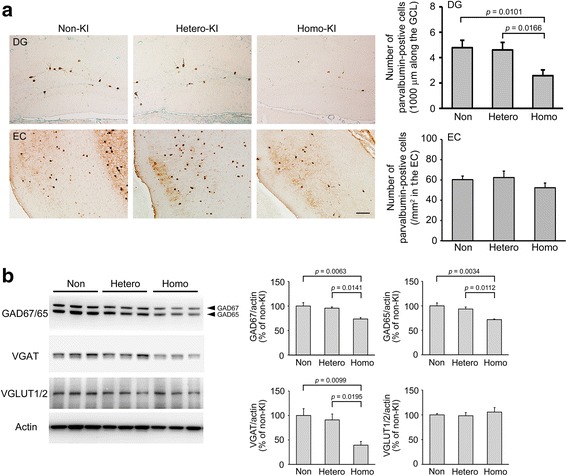
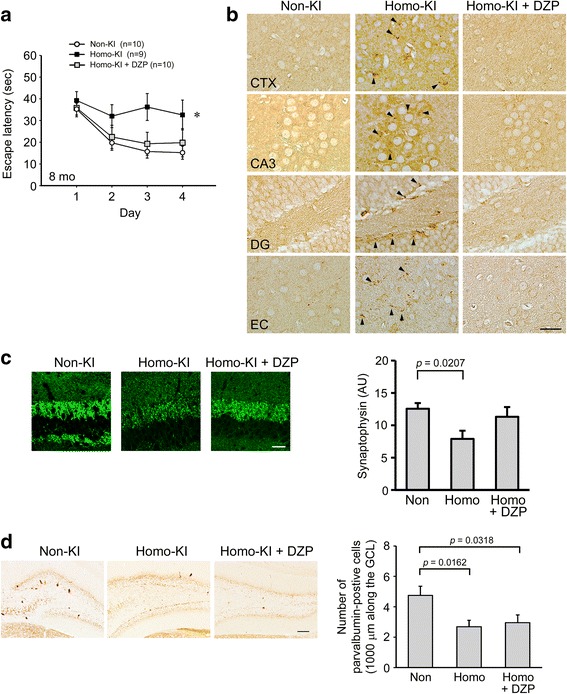
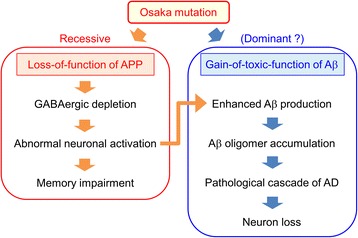
The E693Δ (Osaka) mutation in APP is linked to familial Alzheimer's disease. While this mutation accelerates amyloid β (Aβ) oligomerization, only patient homozygotes suffer from dementia, implying that this mutation is recessive and causes loss-of-function of amyloid precursor protein (APP). To investigate the recessive trait, we generated a new mouse model by knocking-in the Osaka mutation into endogenous mouse APP. The produced homozygous, heterozygous, and non-knockin littermates were compared for memory, neuropathology, and synaptic plasticity. Homozygotes showed memory impairment at 4 months, whereas heterozygotes did not, even at 8 months. Immunohistochemical and biochemical analyses revealed that only homozygotes displayed intraneuronal accumulation of Aβ oligomers at 8 months, followed by abnormal tau phosphorylation, synapse loss, glial activation, and neuron loss. These pathologies were not observed at younger ages, suggesting that a certain mechanism other than Aβ accumulation underlies the memory disturbance at 4 months. For the electrophysiology studies at 4 months, high-frequency stimulation evoked long-term potentiation in all mice in the presence of picrotoxin, but in the absence of picrotoxin, such potentiation was observed only in homozygotes, suggesting their GABAergic deficit. In support of this, the levels of GABA-related proteins and the number of dentate GABAergic interneurons were decreased in 4-month-old homozygotes. Since APP has been shown to play a role in dentate GABAergic synapse formation, the observed GABAergic depletion is likely associated with an impairment of the APP function presumably caused by the Osaka mutation. Oral administration of diazepam to homozygotes from 6 months improved memory at 8 months, and furthermore, prevented Aβ oligomer accumulation, indicating that GABAergic deficiency is a cause of memory impairment and also a driving force of Aβ accumulation. Our findings suggest that the Osaka mutation causes loss of APP function, leading to GABAergic depletion and memory disorder when wild-type APP is absent, providing a mechanism of the recessive heredity.
Keywords: Alzheimer’s disease; GABA; Knockin mouse; Loss of function; Recessive mutation.
Conflict of interest statement
Ethics approval
All procedures performed in studies involving animals were in accordance with the ethical standards of the institution at which the studies were conducted.
Consent for publication
Not applicable.
Competing interests
Drs. Umeda, Yoshida, Morita, Mori, and Tomiyama have a Japanese patent (No. 2015–50,032) on the knockin mouse pending.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
Hypercholesterolemia accelerates intraneuronal accumulation of Aβ oligomers resulting in memory impairment in Alzheimer's disease model mice.Life Sci. 2012 Dec 10;91(23-24):1169-76. doi: 10.1016/j.lfs.2011.12.022. Epub 2012 Jan 17. Life Sci. 2012. PMID: 22273754
-
APP Osaka Mutation in Familial Alzheimer's Disease-Its Discovery, Phenotypes, and Mechanism of Recessive Inheritance.Int J Mol Sci. 2020 Feb 19;21(4):1413. doi: 10.3390/ijms21041413. Int J Mol Sci. 2020. PMID: 32093100 Free PMC article. Review.
-
Neurofibrillary tangle formation by introducing wild-type human tau into APP transgenic mice.Acta Neuropathol. 2014 May;127(5):685-98. doi: 10.1007/s00401-014-1259-1. Epub 2014 Feb 15. Acta Neuropathol. 2014. PMID: 24531886
-
Early accumulation of intracellular fibrillar oligomers and late congophilic amyloid angiopathy in mice expressing the Osaka intra-Aβ APP mutation.Transl Psychiatry. 2012 Nov 13;2(11):e183. doi: 10.1038/tp.2012.109. Transl Psychiatry. 2012. PMID: 23149447 Free PMC article.
-
Alzheimer's Disease Animal Models: Elucidation of Biomarkers and Therapeutic Approaches for Cognitive Impairment.Int J Mol Sci. 2021 May 24;22(11):5549. doi: 10.3390/ijms22115549. Int J Mol Sci. 2021. PMID: 34074018 Free PMC article. Review.
Cited by
-
APP Knock-In Mice Produce E22P-Aβ Exhibiting an Alzheimer's Disease-like Phenotype with Dysregulation of Hypoxia-Inducible Factor Expression.Int J Mol Sci. 2022 Oct 31;23(21):13259. doi: 10.3390/ijms232113259. Int J Mol Sci. 2022. PMID: 36362046 Free PMC article.
-
A bibliometric analysis of the recent advances in diazepam from 2012 to 2021.Front Pharmacol. 2022 Nov 9;13:1042594. doi: 10.3389/fphar.2022.1042594. eCollection 2022. Front Pharmacol. 2022. PMID: 36438847 Free PMC article.
-
Unveiling the therapeutic potential of natural products in Alzheimer's disease: insights from in vitro, in vivo, and clinical studies.Front Pharmacol. 2025 Jun 23;16:1601712. doi: 10.3389/fphar.2025.1601712. eCollection 2025. Front Pharmacol. 2025. PMID: 40626308 Free PMC article. Review.
-
Critical thinking of Alzheimer's transgenic mouse model: current research and future perspective.Sci China Life Sci. 2023 Dec;66(12):2711-2754. doi: 10.1007/s11427-022-2357-x. Epub 2023 Jul 17. Sci China Life Sci. 2023. PMID: 37480469 Review.
-
Glial Contribution to Excitatory and Inhibitory Synapse Loss in Neurodegeneration.Front Cell Neurosci. 2019 Feb 26;13:63. doi: 10.3389/fncel.2019.00063. eCollection 2019. Front Cell Neurosci. 2019. PMID: 30863284 Free PMC article. Review.
References
-
- Chung JK, Nakajima S, Shinagawa S, Plitman E, Chakravarty MM, Iwata Y, Caravaggio F, Pollock BG, Gerretsen P, Graff-Guerrero A, Initiative A’s DN. Benzodiazepine use attenuates cortical β-Amyloid and is not associated with progressive cognitive decline in nondemented elderly adults: a pilot study using F18-Florbetapir positron emission tomography. Am J Geriatr Psychiatry. 2016;24:1028–1039. doi: 10.1016/j.jagp.2016.04.013. - DOI - PubMed
-
- Di Fede G, Catania M, Morbin M, Rossi G, Suardi S, Mazzoleni G, Merlin M, Giovagnoli AR, Prioni S, Erbetta A, Falcone C, Gobbi M, Colombo L, Bastone A, Beeg M, Manzoni C, Francescucci B, Spagnoli A, Cantù L, Del Favero E, Levy E, Salmona M, Tagliavini F. A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science. 2009;323:1473–1477. doi: 10.1126/science.1168979. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
