

Next-generation sequencing to solve complex inherited retinal dystrophy: A case series of multiple genes contributing to disease in extended families
- PMID: 28761320
- PMCID: PMC5524430
Next-generation sequencing to solve complex inherited retinal dystrophy: A case series of multiple genes contributing to disease in extended families
Abstract
Purpose: With recent availability of next-generation sequencing (NGS), it is becoming more common to pursue disease-targeted panel testing rather than traditional sequential gene-by-gene dideoxy sequencing. In this report, we describe using NGS to identify multiple disease-causing mutations that contribute concurrently or independently to retinal dystrophy in three relatively small families.
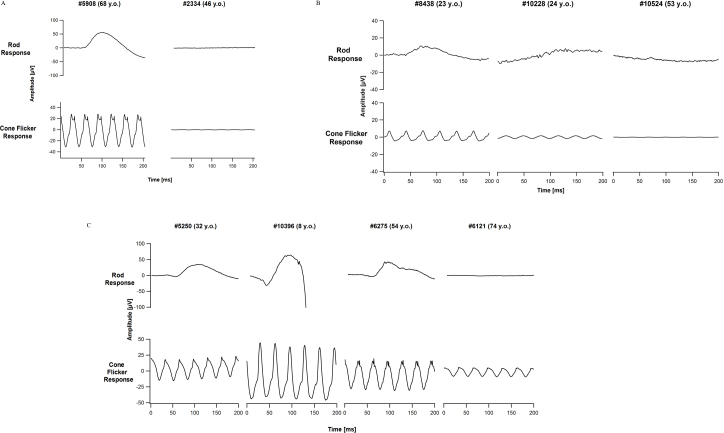
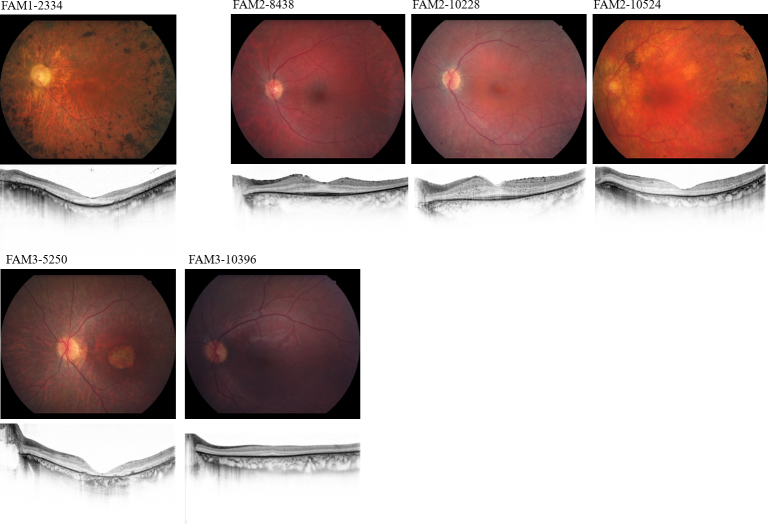
Methods: Family members underwent comprehensive visual function evaluations, and genetic counseling including a detailed family history. A preliminary genetic inheritance pattern was assigned and updated as additional family members were tested. Family 1 (FAM1) and Family 2 (FAM2) were clinically diagnosed with retinitis pigmentosa (RP) and had a suspected autosomal dominant pedigree with non-penetrance (n.p.). Family 3 (FAM3) consisted of a large family with a diagnosis of RP and an overall dominant pedigree, but the proband had phenotypically cone-rod dystrophy. Initial genetic analysis was performed on one family member with traditional Sanger single gene sequencing and/or panel-based testing, and ultimately, retinal gene-targeted NGS was required to identify the underlying cause of disease for individuals within the three families. Results obtained in these families necessitated further genetic and clinical testing of additional family members to determine the complex genetic and phenotypic etiology of each family.
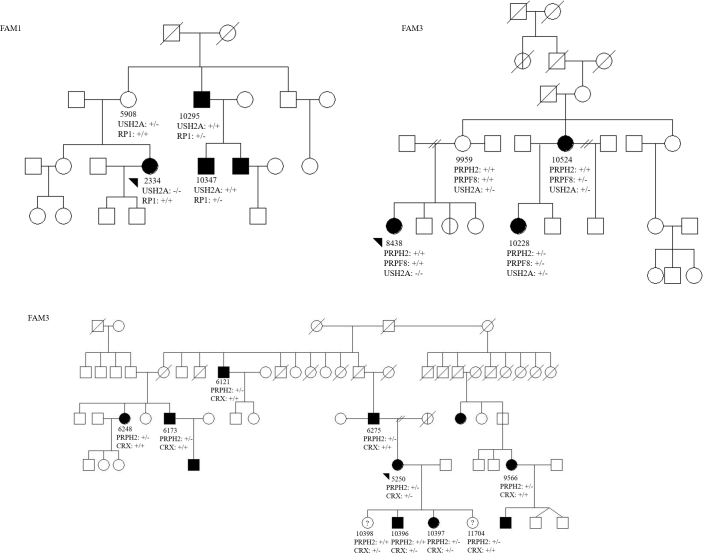
Results: Genetic testing of FAM1 (n = 4 affected; 1 n.p.) identified a dominant mutation in RP1 (p.Arg677Ter) that was present for two of the four affected individuals but absent in the proband and the presumed non-penetrant individual. Retinal gene-targeted NGS in the fourth affected family member revealed compound heterozygous mutations in USH2A (p. Cys419Phe, p.Glu767Serfs*21). Genetic testing of FAM2 (n = 3 affected; 1 n.p.) identified three retinal dystrophy genes (PRPH2, PRPF8, and USH2A) with disease-causing mutations in varying combinations among the affected family members. Genetic testing of FAM3 (n = 7 affected) identified a mutation in PRPH2 (p.Pro216Leu) tracking with disease in six of the seven affected individuals. Additional retinal gene-targeted NGS testing determined that the proband also harbored a multiple exon deletion in the CRX gene likely accounting for her cone-rod phenotype; her son harbored only the mutation in CRX, not the familial mutation in PRPH2.
Conclusions: Multiple genes contributing to the retinal dystrophy genotypes within a family were discovered using retinal gene-targeted NGS. Families with noted examples of phenotypic variation or apparent non-penetrant individuals may offer a clue to suspect complex inheritance. Furthermore, this finding underscores that caution should be taken when attributing a single gene disease-causing mutation (or inheritance pattern) to a family as a whole. Identification of a disease-causing mutation in a proband, even with a clear inheritance pattern in hand, may not be sufficient for targeted, known mutation analysis in other family members.
Figures
References
-
- Haim M. Epidemiology of retinitis pigmentosa in Denmark. Acta Ophthalmol Scand Suppl. 2002;233:1–34. - PubMed
