

Prevalence of spinocerebellar ataxia 36 in a US population
- PMID: 28761930
- PMCID: PMC5515602
- DOI: 10.1212/NXG.0000000000000174
Prevalence of spinocerebellar ataxia 36 in a US population
Abstract
Objective: To assess the prevalence and clinical features of individuals affected by spinocerebellar ataxia 36 (SCA36) at a large tertiary referral center in the United States.
Methods: A total of 577 patients with undiagnosed sporadic or familial cerebellar ataxia comprehensively evaluated at a tertiary referral ataxia center were molecularly evaluated for SCA36. Repeat primed PCR and fragment analysis were used to screen for the presence of a repeat expansion in the NOP56 gene.
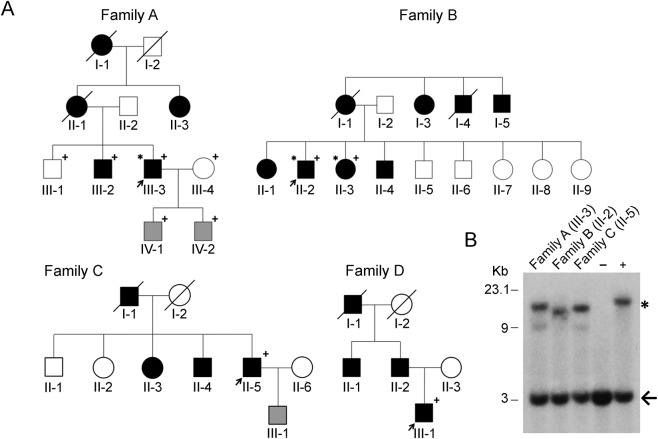
Results: Fragment analysis of triplet repeat primed PCR products identified a GGCCTG hexanucleotide repeat expansion in intron 1 of NOP56 in 4 index cases. These 4 SCA36-positive families comprised 2 distinct ethnic groups: white (European) (2) and Asian (Japanese [1] and Vietnamese [1]). Individuals affected by SCA36 exhibited typical clinical features with gait ataxia and age at onset ranging between 35 and 50 years. Patients also suffered from ataxic or spastic limbs, altered reflexes, abnormal ocular movement, and cognitive impairment.
Conclusions: In a US population, SCA36 was observed to be a rare disorder, accounting for 0.7% (4/577 index cases) of disease in a large undiagnosed ataxia cohort.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources