

Late diagnosis and atypical brain imaging of Aicardi-Goutières syndrome: are we failing to diagnose Aicardi-Goutières syndrome-2?
- PMID: 28762473
- PMCID: PMC5685901
- DOI: 10.1111/dmcn.13509
Late diagnosis and atypical brain imaging of Aicardi-Goutières syndrome: are we failing to diagnose Aicardi-Goutières syndrome-2?
Abstract
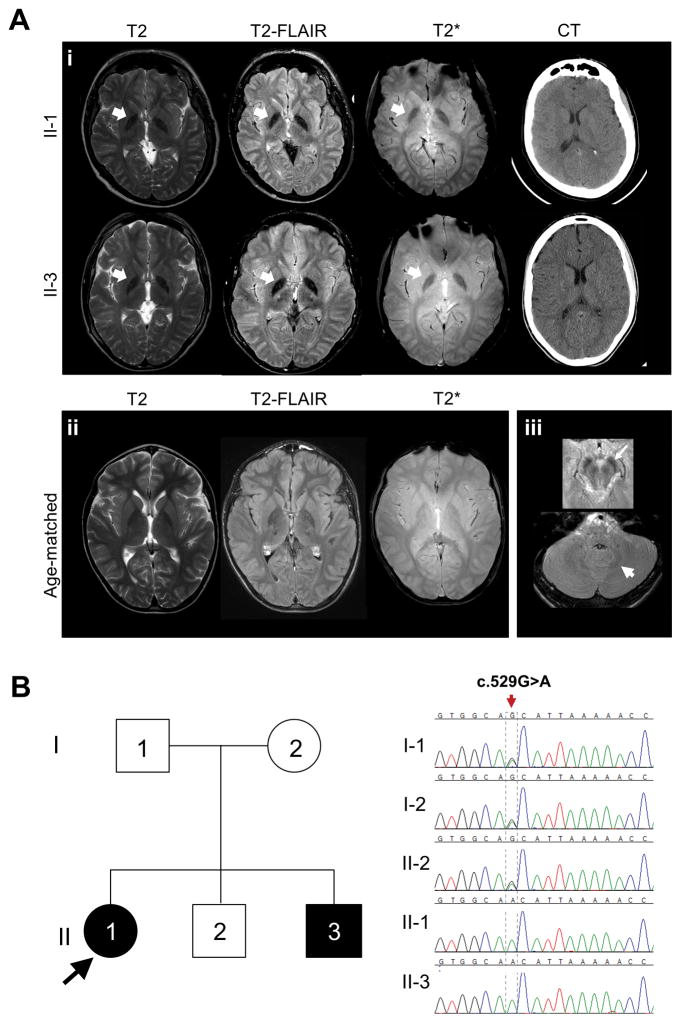
Aicardi-Goutières syndrome (AGS) is a rare disorder with in utero or postnatal onset of encephalopathy and progressive neurological deterioration. The seven genetic subtypes of AGS are associated with abnormal type I interferon-mediated innate immune response. Most patients with AGS present with progressive microcephaly, spasticity, and cognitive impairment. Some, especially those with type 2 (AGS2), manifest milder phenotypes, reduced childhood mortality, and relative preservation of physical and cognitive abilities. In this report, we describe two siblings (sister and brother) diagnosed with AGS2 in their second decade, who exhibited static encephalopathy since 1 year of age with spastic quadriplegia and anarthria but preserved intellect. Both were homozygous for the common pathogenic RNASEH2B allele (c.529G>A, p.Ala177Thr). Rather than manifesting calcifications and leukoencephalopathy, both had increased iron signal in the basal ganglia. Our report broadens the clinical and imaging spectrum of AGS2 and emphasizes the importance of including AGS2 in the differential diagnosis of idiopathic spastic cerebral palsy.
What this paper adds: We identified two siblings (sister and brother) with atypical Aicardi-Goutières syndrome type 2 due to RNASEH2B mutation. Manifestations included spastic quadriplegia and anarthria but preserved intellect and increased iron signal in the basal ganglia. RNASEH2B-related Aicardi-Goutières syndrome type 2 can have present with a variable phenotype, including idiopathic spastic cerebral palsy.
Published 2017. This article is a U.S. Government work and is in the public domain in the USA.
Figures
References
-
- Aicardi J, Goutieres F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol. 1984;15:49–54. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
