

Ensemblator v3: Robust atom-level comparative analyses and classification of protein structure ensembles
- PMID: 28762605
- PMCID: PMC5734391
- DOI: 10.1002/pro.3249
Ensemblator v3: Robust atom-level comparative analyses and classification of protein structure ensembles
Abstract
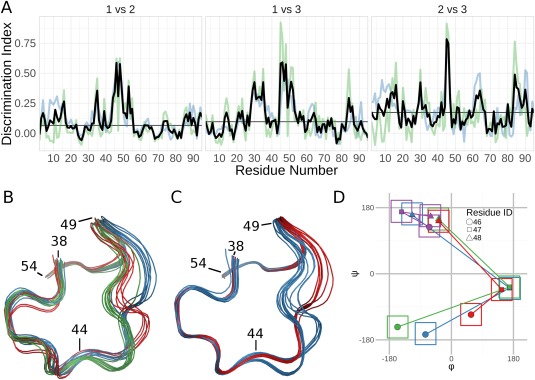
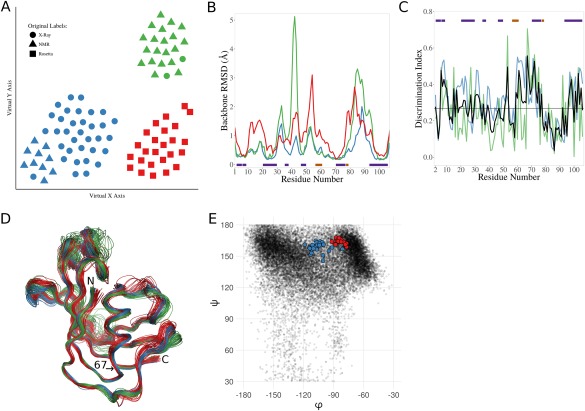
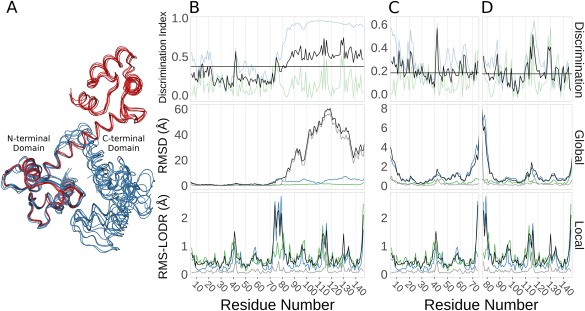
Ensembles of protein structures are increasingly used to represent the conformational variation of a protein as determined by experiment and/or by molecular simulations, as well as uncertainties that may be associated with structure determinations or predictions. Making the best use of such information requires the ability to quantitatively compare entire ensembles. For this reason, we recently introduced the Ensemblator (Clark et al., Protein Sci 2015; 24:1528), a novel approach to compare user-defined groups of models, in residue level detail. Here we describe Ensemblator v3, an open-source program that employs the same basic ensemble comparison strategy but includes major advances that make it more robust, powerful, and user-friendly. Ensemblator v3 carries out multiple sequence alignments to facilitate the generation of ensembles from non-identical input structures, automatically optimizes the key global overlay parameter, optionally performs "ensemble clustering" to classify the models into subgroups, and calculates a novel "discrimination index" that quantifies similarities and differences, at residue or atom level, between each pair of subgroups. The clustering and automatic options mean that no pre-knowledge about an ensemble is required for its analysis. After describing the novel features of Ensemblator v3, we demonstrate its utility using three case studies that illustrate the ease with which complex analyses are accomplished, and the kinds of insights derived from clustering into subgroups and from the detailed information that locates significant differences. The Ensemblator v3 enhances the structural biology toolbox by greatly expanding the kinds of problems to which this ensemble comparison strategy can be applied.
Keywords: NMR ensemble; Rosetta; clustering; ensemble clustering; protein structure comparison; python; structure prediction; superposition; template-based modeling.
© 2017 The Protein Society.
Figures
Similar articles
-
Residue-level global and local ensemble-ensemble comparisons of protein domains.Protein Sci. 2015 Sep;24(9):1528-42. doi: 10.1002/pro.2714. Epub 2015 Jun 22. Protein Sci. 2015. PMID: 26032515 Free PMC article.
-
ENCORE: Software for Quantitative Ensemble Comparison.PLoS Comput Biol. 2015 Oct 27;11(10):e1004415. doi: 10.1371/journal.pcbi.1004415. eCollection 2015 Oct. PLoS Comput Biol. 2015. PMID: 26505632 Free PMC article.
-
Exploring the extremes of sequence/structure space with ensemble fold recognition in the program Phyre.Proteins. 2008 Feb 15;70(3):611-25. doi: 10.1002/prot.21688. Proteins. 2008. PMID: 17876813
-
The Protein Ensemble Database.Adv Exp Med Biol. 2015;870:335-49. doi: 10.1007/978-3-319-20164-1_11. Adv Exp Med Biol. 2015. PMID: 26387108 Review.
-
Ensemble-function relationships: From qualitative to quantitative relationships between protein structure and function.J Struct Biol. 2025 Mar;217(1):108152. doi: 10.1016/j.jsb.2024.108152. Epub 2024 Dec 9. J Struct Biol. 2025. PMID: 39577782 Review.
Cited by
-
Proteomimetic Zinc Finger Domains with Modified Metal-binding β-Turns.Pept Sci (Hoboken). 2020 Sep;112(5):e24177. doi: 10.1002/pep2.24177. Epub 2020 Jun 7. Pept Sci (Hoboken). 2020. PMID: 33733039 Free PMC article.
-
Systematic identification of recognition motifs for the hub protein LC8.Life Sci Alliance. 2019 Jul 2;2(4):e201900366. doi: 10.26508/lsa.201900366. Print 2019 Aug. Life Sci Alliance. 2019. PMID: 31266884 Free PMC article.
-
Automated NMR resonance assignments and structure determination using a minimal set of 4D spectra.Nat Commun. 2018 Jan 26;9(1):384. doi: 10.1038/s41467-017-02592-z. Nat Commun. 2018. PMID: 29374165 Free PMC article.
-
Ligand-induced shifts in conformational ensembles that describe transcriptional activation.Elife. 2022 Oct 12;11:e80140. doi: 10.7554/eLife.80140. Elife. 2022. PMID: 36222302 Free PMC article.
-
Heterogeneous-Backbone Proteomimetic Analogues of Lasiocepsin, a Disulfide-Rich Antimicrobial Peptide with a Compact Tertiary Fold.ACS Chem Biol. 2022 Apr 15;17(4):987-997. doi: 10.1021/acschembio.2c00138. Epub 2022 Mar 15. ACS Chem Biol. 2022. PMID: 35290019 Free PMC article.
References
-
- Elber R, Karplus M (1987) Multiple conformational states of proteins: a molecular dynamics analysis of myoglobin. Science 235:318–321. - PubMed
-
- Furnham N, Blundell TL, DePristo MA, Terwilliger TC (2006) Is one solution good enough? Nat Struct Mol Biol 13:184–185. - PubMed
-
- Lindorff‐Larsen K, Best RB, DePristo MA, Dobson CM, Vendruscolo M (2005) Simultaneous determination of protein structure and dynamics. Nature 433:128–132. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
