

Synthesis of novel coumarin nucleus-based DPA drug-like molecular entity: In vitro DNA/Cu(II) binding, DNA cleavage and pro-oxidant mechanism for anticancer action
- PMID: 28763458
- PMCID: PMC5538679
- DOI: 10.1371/journal.pone.0181783
Synthesis of novel coumarin nucleus-based DPA drug-like molecular entity: In vitro DNA/Cu(II) binding, DNA cleavage and pro-oxidant mechanism for anticancer action
Abstract
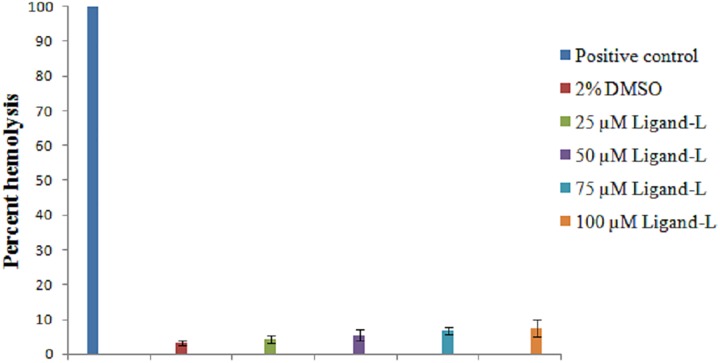
Despite substantial research on cancer therapeutics, systemic toxicity and drug-resistance limits the clinical application of many drugs like cisplatin. Therefore, new chemotherapeutic strategies against different malignancies are needed. Targeted cancer therapy is a new paradigm for cancer therapeutics which targets pathways or chemical entities specific to cancer cells than normal ones. Unlike normal cells, cancer cells contain elevated copper which plays an integral role in angiogenesis. Copper is an important metal ion associated with chromatin DNA, particularly with guanine. Thus, targeting copper via copper-specific chelators in cancer cells can serve as an effective anticancer strategy. New pharmacophore di(2-picolyl)amine (DPA)-3(bromoacetyl) coumarin (ligand-L) was synthesized and characterized by IR, ESI-MS, 1H- and 13C-NMR. Binding ability of ligand-L to DNA/Cu(II) was evaluated using a plethora of biophysical techniques which revealed ligand-L-DNA and ligand-L-Cu(II) interaction. Competitive displacement assay and docking confirmed non-intercalative binding mode of ligand-L with ctDNA. Cyclic voltammetry confirmed ligand-L causes quasi reversible Cu(II)/Cu(I) conversion. Further, acute toxicity studies revealed no toxic effects of ligand-L on mice. To evaluate the chemotherapeutic potential and anticancer mechanism of ligand-L, DNA damage via pBR322 cleavage assay and reactive oxygen species (ROS) generation were studied. Results demonstrate that ligand-L causes DNA cleavage involving ROS generation in the presence of Cu(II). In conclusion, ligand-L causes redox cycling of Cu(II) to generate ROS which leads to oxidative DNA damage and pro-oxidant cancer cell death. These findings will establish ligand-L as a lead molecule to synthesize new molecules with better copper chelating and pro-oxidant properties against different malignancies.
Conflict of interest statement
Figures














References
-
- Hurley LH. DNA and its associated processes as targets for cancer therapy. Nat Rev Cancer. 2002; 2:188–200. doi: 10.1038/nrc749 - DOI - PubMed
-
- Burger RM. Cleavage of nucleic acids by bleomycin. Chem Rev. 1998; 98:1153–1170. - PubMed
-
- Boerner LJ, Zaleski JM. Metal complex-DNA interactions: from transcription inhibition to photoactivated cleavage. Curr Opin Chem Biol. 2005; 9:135–144. doi: 10.1016/j.cbpa.2005.02.010 - DOI - PubMed
-
- Zhang CX, Lippard SJ. New metal complexes as potential therapeutics. Curr Opin Chem Biol. 2003; 7:481–489. - PubMed
-
- Boulikas T, Vougiouka M. Cisplatin and platinum drugs at the molecular level. (Review). Oncol Rep. 2003; 10:1663–1682. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
