

CDHR1 mutations in retinal dystrophies
- PMID: 28765526
- PMCID: PMC5539332
- DOI: 10.1038/s41598-017-07117-8
CDHR1 mutations in retinal dystrophies
Abstract
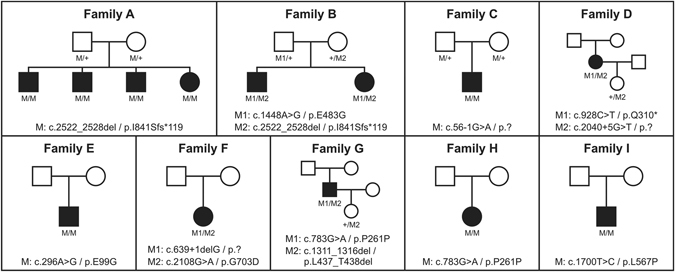
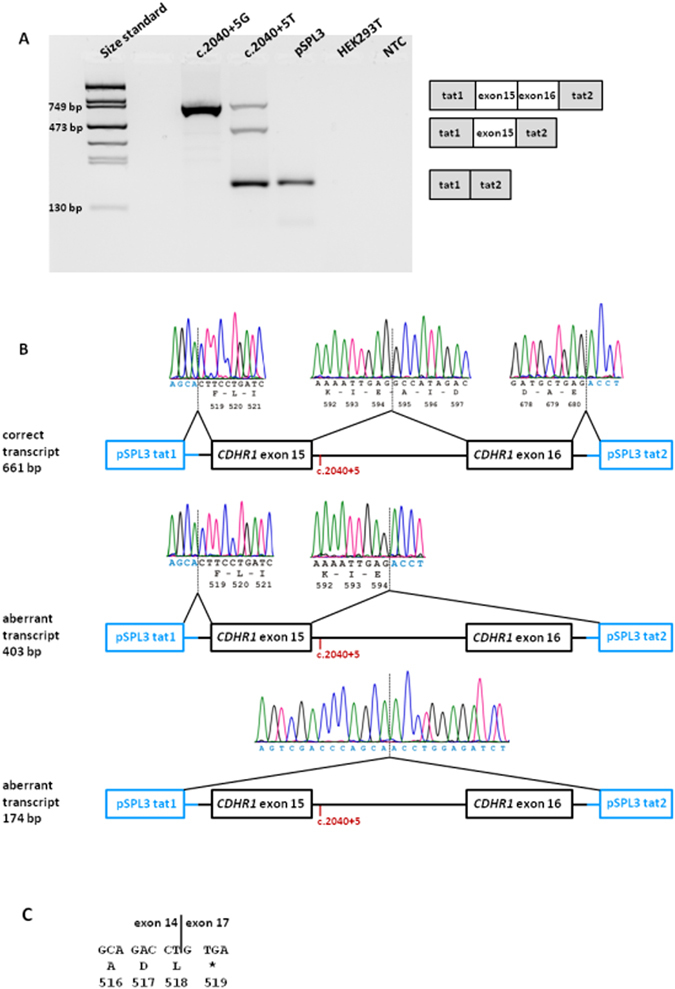
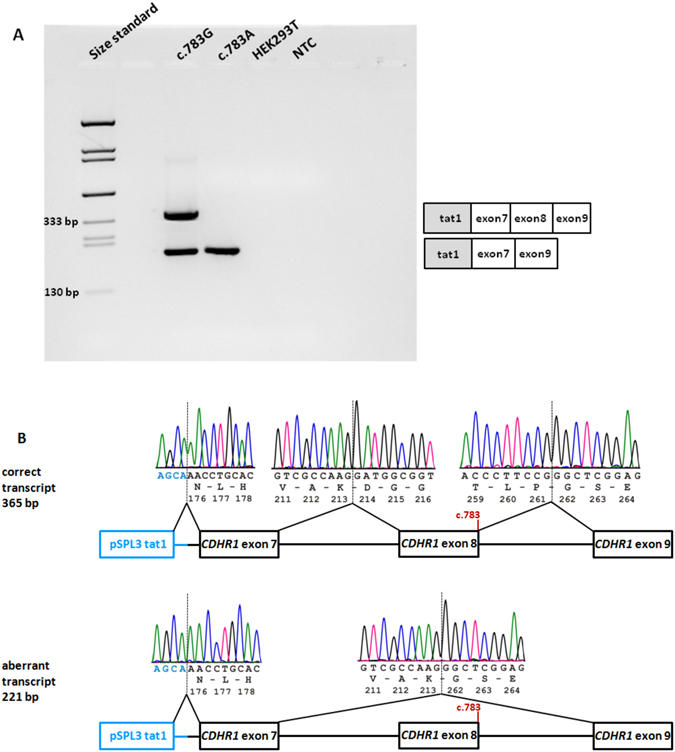
We report ophthalmic and genetic findings in patients with autosomal recessive retinitis pigmentosa (RP), cone-rod dystrophy (CRD) or cone dystrophy (CD) harboring potential pathogenic variants in the CDHR1 gene. Detailed ophthalmic examination was performed in seven sporadic and six familial subjects. Mutation screening was done using a customized next generation sequencing panel targeting 105 genes implicated in inherited retinal disorders. In one family, homozygosity mapping with subsequent candidate gene analysis was performed. Stringent filtering for rare and potentially disease causing variants following a model of autosomal recessive inheritance led to the identification of eleven different CDHR1 variants in nine index cases. All variants were novel at the time of their identification. In silico analyses confirmed their pathogenic potential. Minigene assays were performed for two non-canonical splice site variants and revealed missplicing for the mutant alleles. Mutations in CDHR1 are a rare cause of retinal dystrophy. Our study further expands the mutational spectrum of this gene and the associated clinical presentation.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
-
- Weleber, R. G. Retinitis pigmentosa and allied disorders in Retina (ed. Ryan, S. J.) 335–466 (Mosby, 1994).
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
