

APP mouse models for Alzheimer's disease preclinical studies
- PMID: 28768718
- PMCID: PMC5579350
- DOI: 10.15252/embj.201797397
APP mouse models for Alzheimer's disease preclinical studies
Abstract
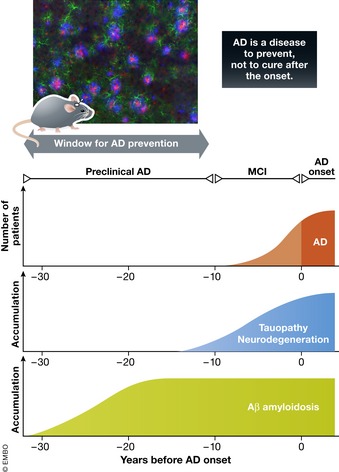
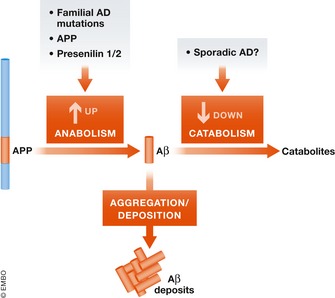
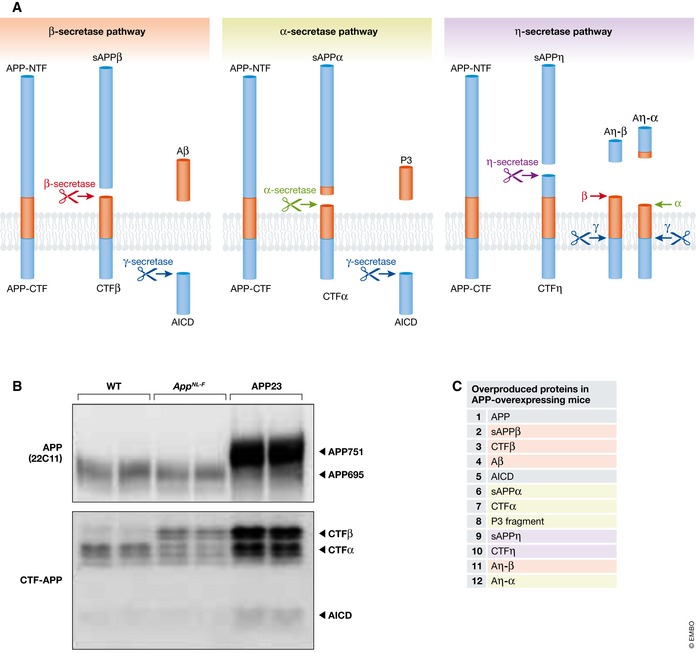
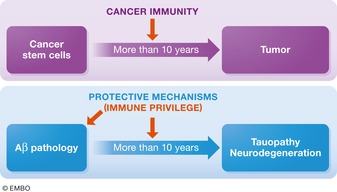
Animal models of human diseases that accurately recapitulate clinical pathology are indispensable for understanding molecular mechanisms and advancing preclinical studies. The Alzheimer's disease (AD) research community has historically used first-generation transgenic (Tg) mouse models that overexpress proteins linked to familial AD (FAD), mutant amyloid precursor protein (APP), or APP and presenilin (PS). These mice exhibit AD pathology, but the overexpression paradigm may cause additional phenotypes unrelated to AD Second-generation mouse models contain humanized sequences and clinical mutations in the endogenous mouse App gene. These mice show Aβ accumulation without phenotypes related to overexpression but are not yet a clinical recapitulation of human AD In this review, we evaluate different APP mouse models of AD, and review recent studies using the second-generation mice. We advise AD researchers to consider the comparative strengths and limitations of each model against the scientific and therapeutic goal of a prospective preclinical study.
Keywords: APP transgenic; Alzheimer's disease; App knock‐in; amyloid precursor protein; amyloid β peptide.
© 2017 The Authors. Published under the terms of the CC BY 4.0 license.
Figures
References
-
- Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN et al (2012) Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med 367: 795–804 - PMC - PubMed
-
- Benilova I, Gallardo R, Ungureanu AA, Castillo Cano V, Snellinx A, Ramakers M, Bartic C, Rousseau F, Schymkowitz J, De Strooper B (2014) The Alzheimer disease protective mutation A2T modulates kinetic and thermodynamic properties of amyloid‐β (Aβ) aggregation. J Biol Chem 289: 30977–30989 - PMC - PubMed
-
- Bolmont T, Clavaguera F, Meyer‐Luehmann M, Herzig MC, Radde R, Staufenbiel M, Lewis J, Hutton M, Tolnay M, Jucker M (2007) Induction of tau pathology by intracerebral infusion of amyloid‐β‐containing brain extract and by amyloid‐beta deposition in APP x Tau transgenic mice. Am J Pathol 171: 2012–2020 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
