

Nonmyocyte ERK1/2 signaling contributes to load-induced cardiomyopathy in Marfan mice
- PMID: 28768908
- PMCID: PMC5543913
- DOI: 10.1172/jci.insight.91588
Nonmyocyte ERK1/2 signaling contributes to load-induced cardiomyopathy in Marfan mice
Abstract
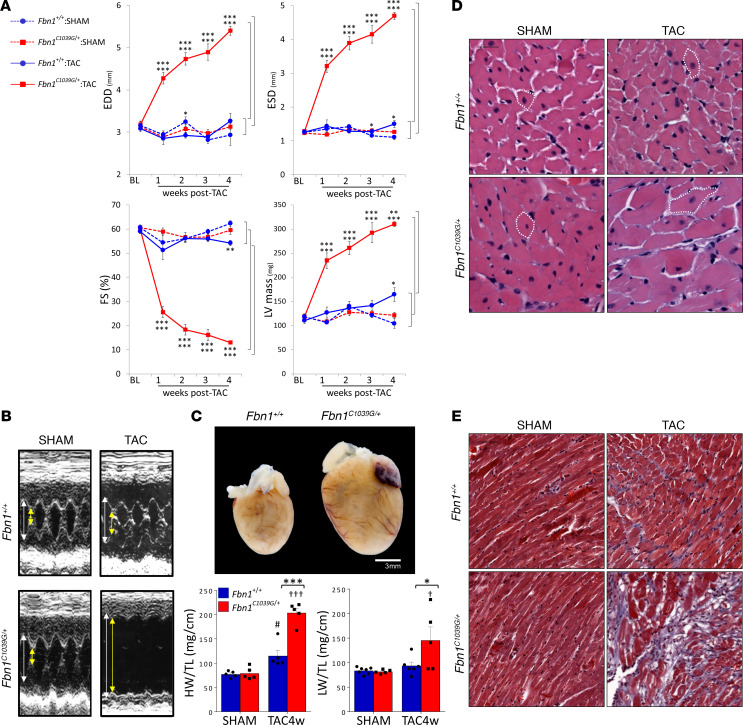
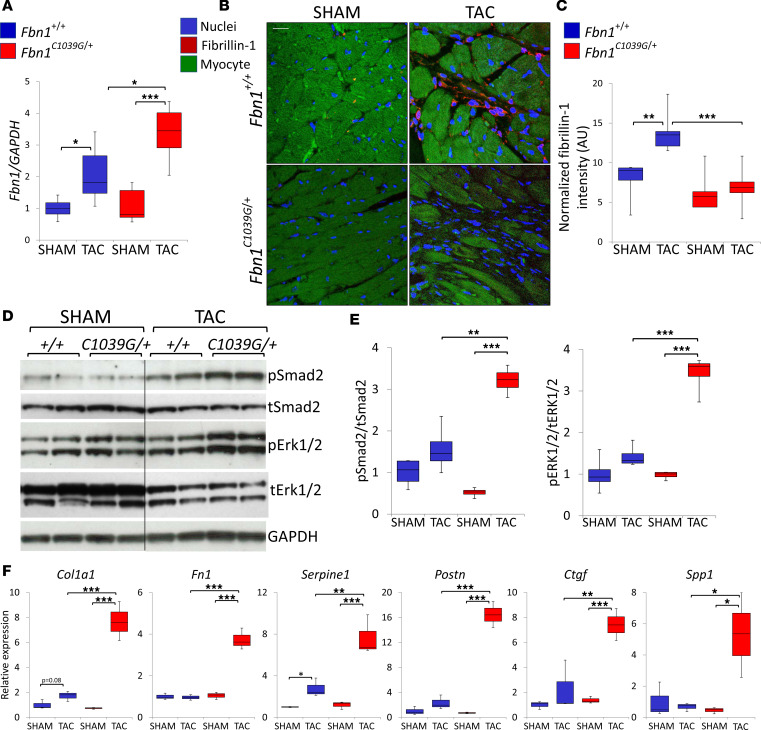
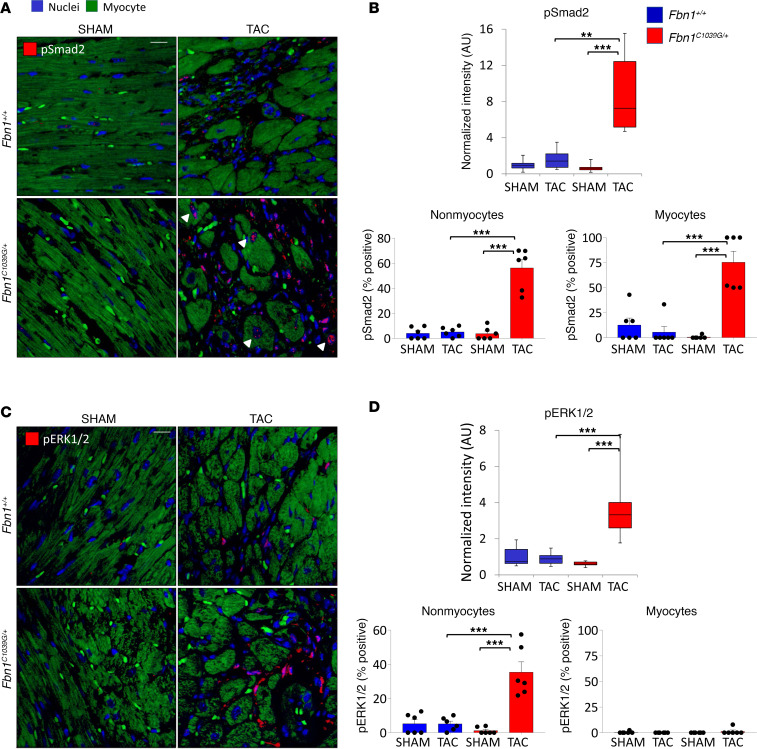
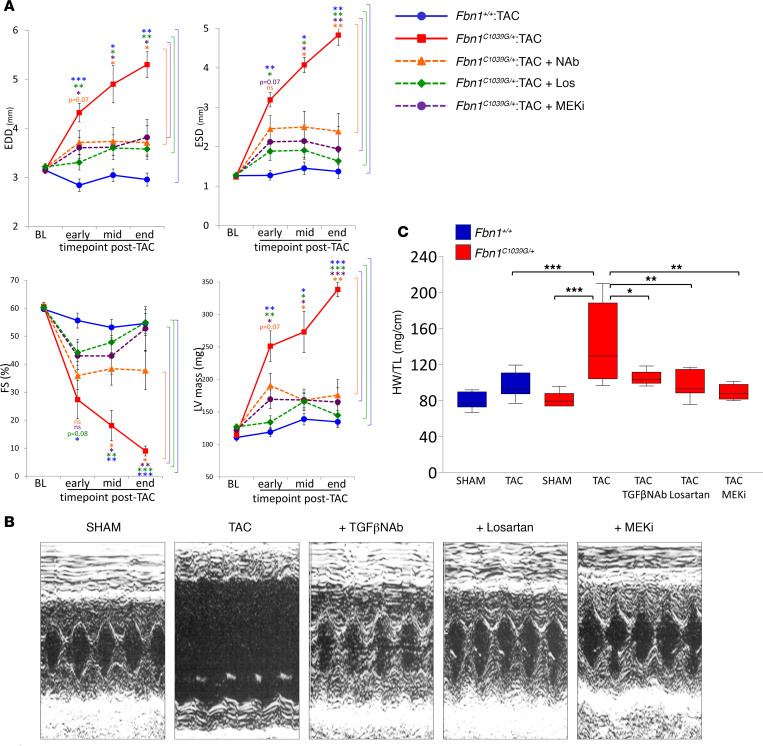
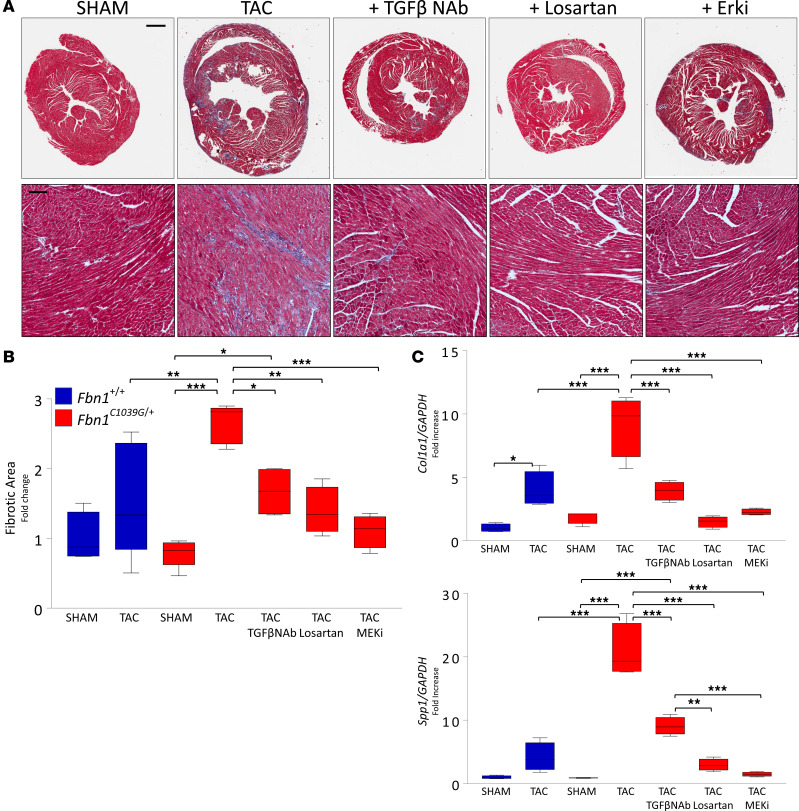
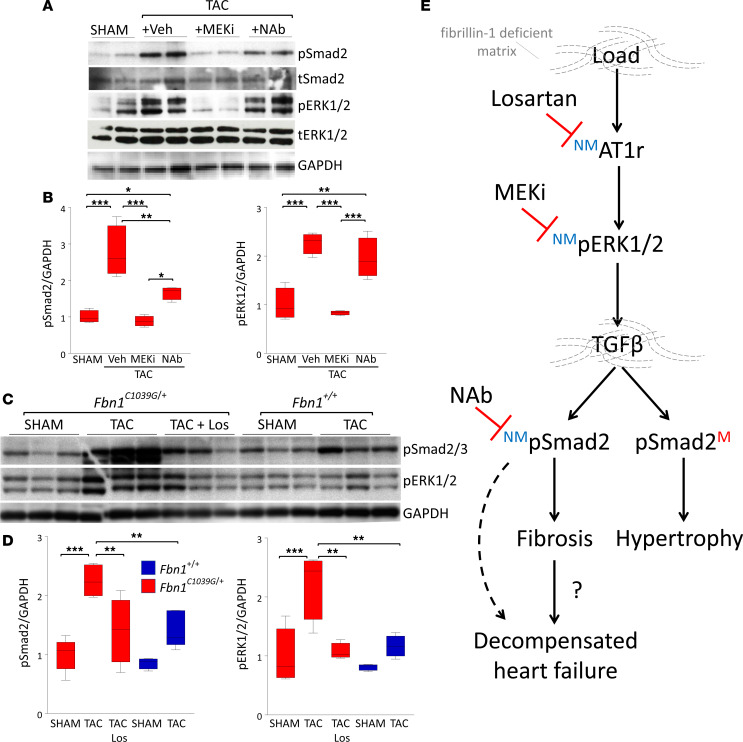
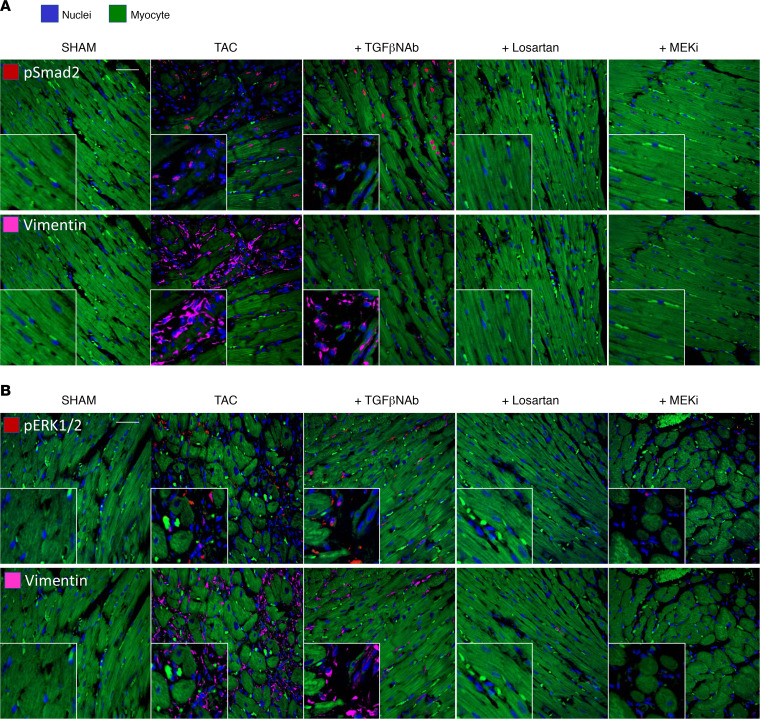
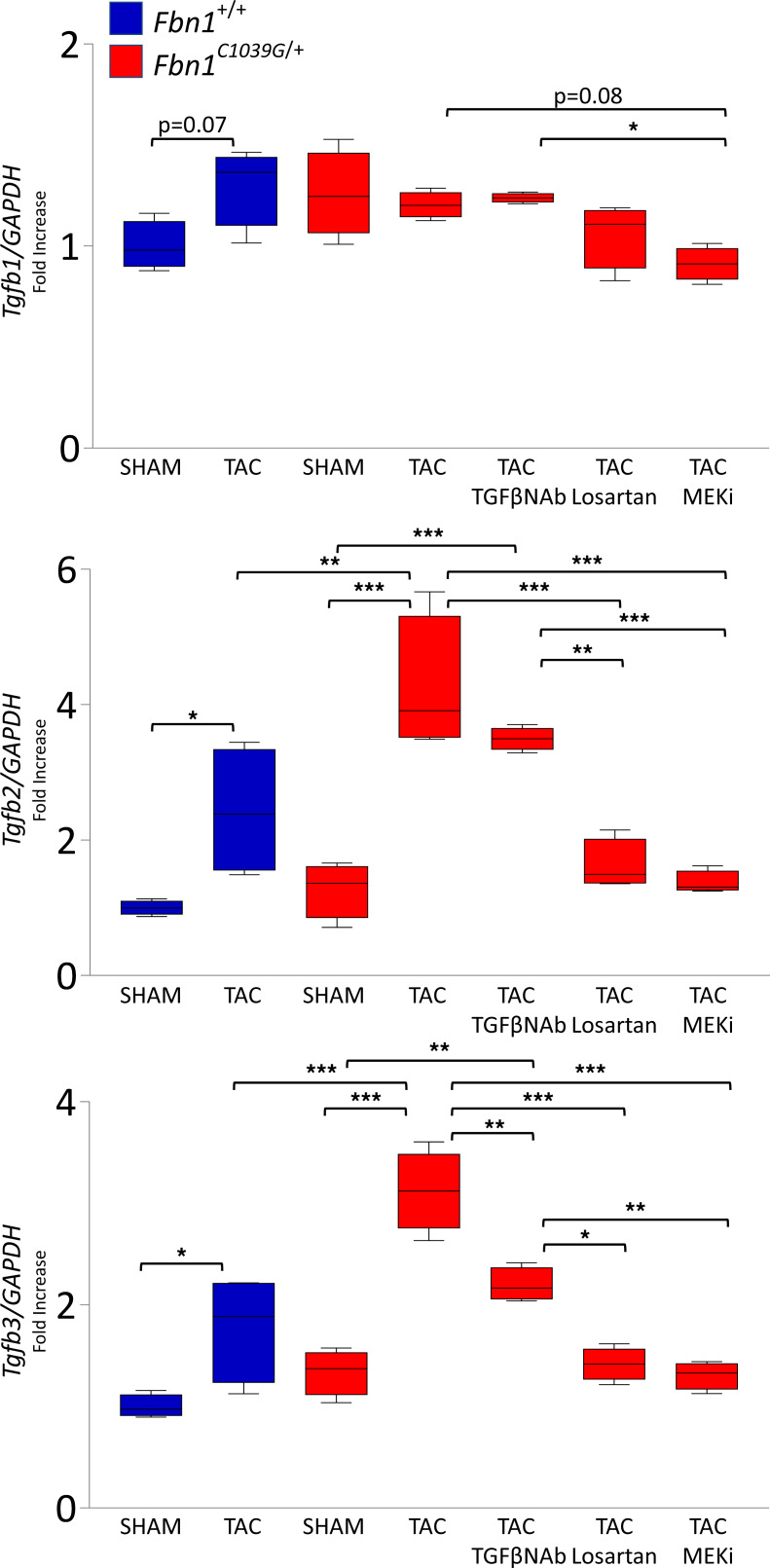
Among children with the most severe presentation of Marfan syndrome (MFS), an inherited disorder of connective tissue caused by a deficiency of extracellular fibrillin-1, heart failure is the leading cause of death. Here, we show that, while MFS mice (Fbn1C1039G/+ mice) typically have normal cardiac function, pressure overload (PO) induces an acute and severe dilated cardiomyopathy in association with fibrosis and myocyte enlargement. Failing MFS hearts show high expression of TGF-β ligands, with increased TGF-β signaling in both nonmyocytes and myocytes; pathologic ERK activation is restricted to the nonmyocyte compartment. Informatively, TGF-β, angiotensin II type 1 receptor (AT1R), or ERK antagonism (with neutralizing antibody, losartan, or MEK inhibitor, respectively) prevents load-induced cardiac decompensation in MFS mice, despite persistent PO. In situ analyses revealed an unanticipated axis of activation in nonmyocytes, with AT1R-dependent ERK activation driving TGF-β ligand expression that culminates in both autocrine and paracrine overdrive of TGF-β signaling. The full compensation seen in wild-type mice exposed to mild PO correlates with enhanced deposition of extracellular fibrillin-1. Taken together, these data suggest that fibrillin-1 contributes to cardiac reserve in the face of hemodynamic stress, critically implicate nonmyocytes in disease pathogenesis, and validate ERK as a therapeutic target in MFS-related cardiac decompensation.
Keywords: Cardiology; Genetics.
Conflict of interest statement
Figures
References
-
- Morse RP, et al. Diagnosis and management of infantile Marfan syndrome. Pediatrics. 1990;86(6):888–895. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
