

Unraveling Core Functional Microbiota in Traditional Solid-State Fermentation by High-Throughput Amplicons and Metatranscriptomics Sequencing
- PMID: 28769888
- PMCID: PMC5509801
- DOI: 10.3389/fmicb.2017.01294
Unraveling Core Functional Microbiota in Traditional Solid-State Fermentation by High-Throughput Amplicons and Metatranscriptomics Sequencing
Abstract
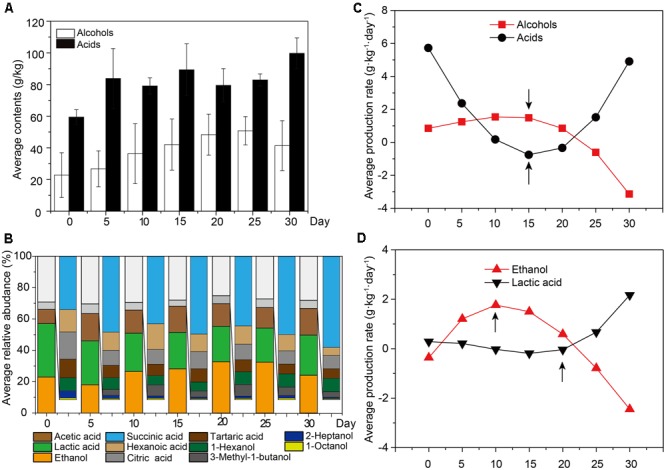
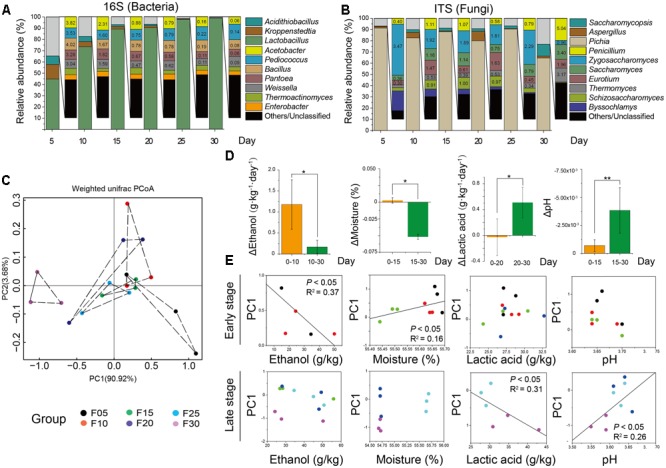
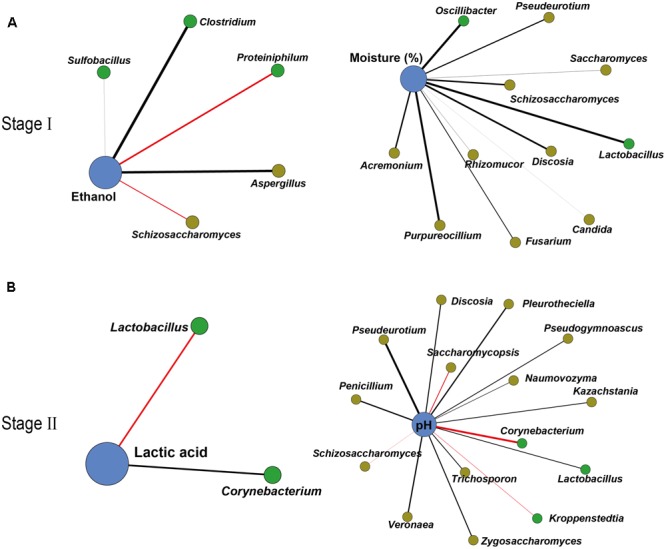
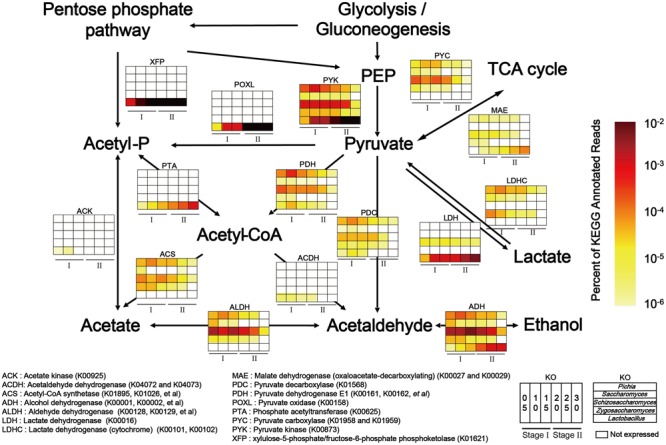
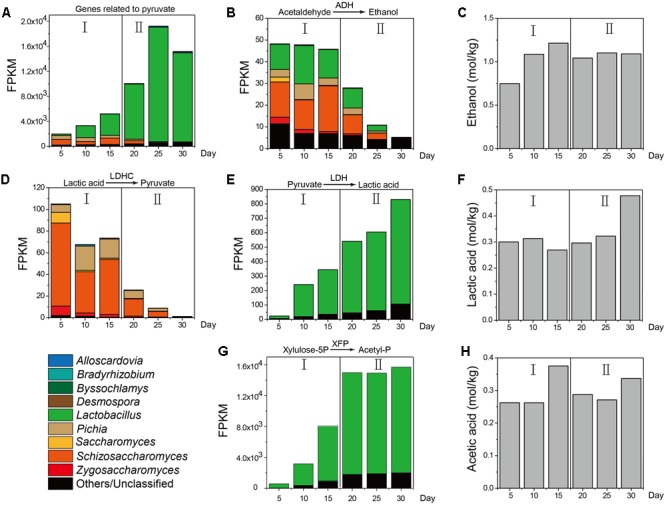
Fermentation microbiota is specific microorganisms that generate different types of metabolites in many productions. In traditional solid-state fermentation, the structural composition and functional capacity of the core microbiota determine the quality and quantity of products. As a typical example of food fermentation, Chinese Maotai-flavor liquor production involves a complex of various microorganisms and a wide variety of metabolites. However, the microbial succession and functional shift of the core microbiota in this traditional food fermentation remain unclear. Here, high-throughput amplicons (16S rRNA gene amplicon sequencing and internal transcribed space amplicon sequencing) and metatranscriptomics sequencing technologies were combined to reveal the structure and function of the core microbiota in Chinese soy sauce aroma type liquor production. In addition, ultra-performance liquid chromatography and headspace-solid phase microextraction-gas chromatography-mass spectrometry were employed to provide qualitative and quantitative analysis of the major flavor metabolites. A total of 10 fungal and 11 bacterial genera were identified as the core microbiota. In addition, metatranscriptomic analysis revealed pyruvate metabolism in yeasts (genera Pichia, Schizosaccharomyces, Saccharomyces, and Zygosaccharomyces) and lactic acid bacteria (genus Lactobacillus) classified into two stages in the production of flavor components. Stage I involved high-level alcohol (ethanol) production, with the genus Schizosaccharomyces serving as the core functional microorganism. Stage II involved high-level acid (lactic acid and acetic acid) production, with the genus Lactobacillus serving as the core functional microorganism. The functional shift from the genus Schizosaccharomyces to the genus Lactobacillus drives flavor component conversion from alcohol (ethanol) to acid (lactic acid and acetic acid) in Chinese Maotai-flavor liquor production. Our findings provide insight into the effects of the core functional microbiota in soy sauce aroma type liquor production and the characteristics of the fermentation microbiota under different environmental conditions.
Keywords: amplicons; core functional microbiota; fermentation microbiota; functional shift; high-throughput sequencing; metatranscriptomics; microbial succession; solid-state fermentation.
Figures
References
-
- Azwar M. Y., Hussain M. A., Abdul-Wahab A. K. (2014). Development of biohydrogen production by photobiological, fermentation and electrochemical processes: a review. Renew. Sustain. Energy Rev. 31 158–173. 10.1016/j.rser.2013.11.022 - DOI
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials