

Reprogramming of Tumor-Associated Macrophages with Anticancer Therapies: Radiotherapy versus Chemo- and Immunotherapies
- PMID: 28769933
- PMCID: PMC5509958
- DOI: 10.3389/fimmu.2017.00828
Reprogramming of Tumor-Associated Macrophages with Anticancer Therapies: Radiotherapy versus Chemo- and Immunotherapies
Abstract
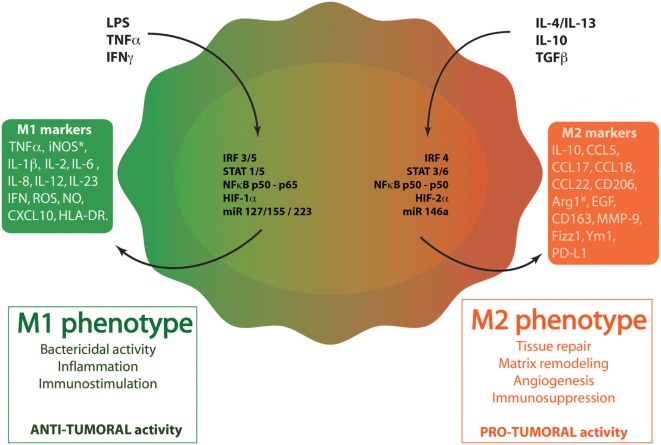
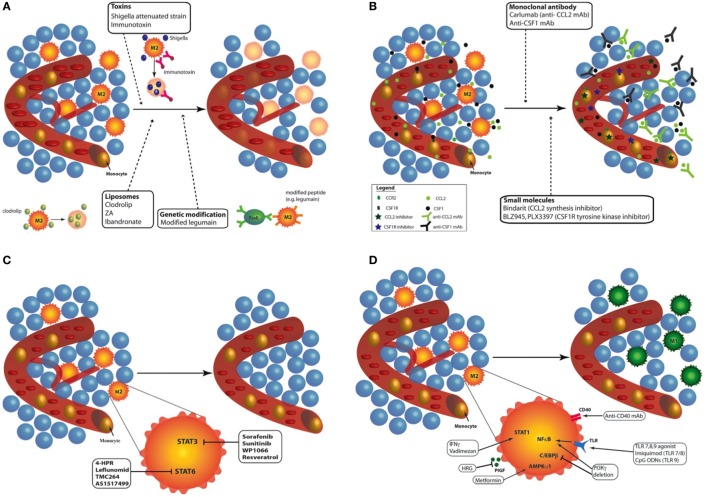
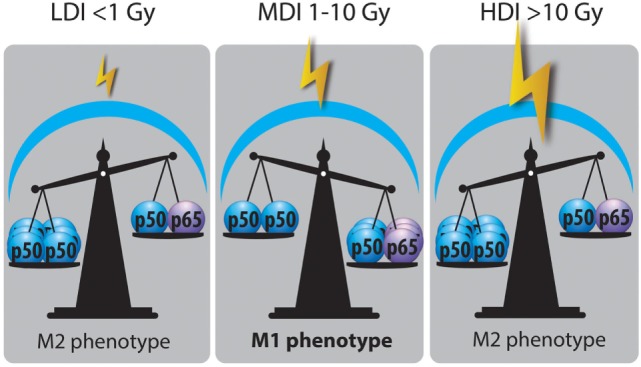
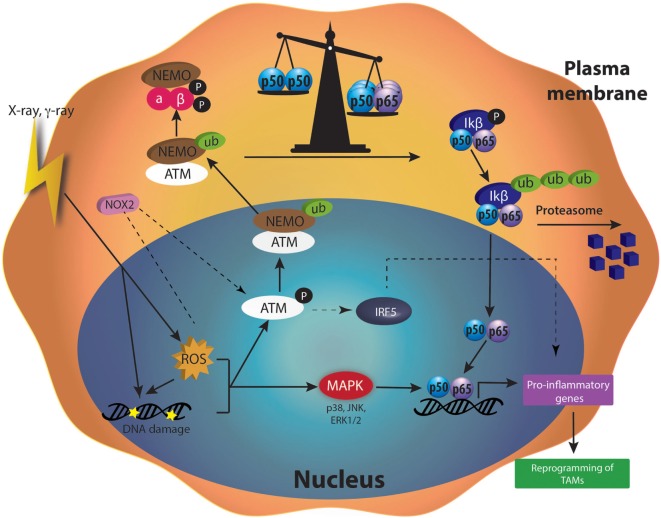
Tumor-associated macrophages (TAMs) play a central role in tumor progression, metastasis, and recurrence after treatment. Macrophage plasticity and diversity allow their classification along a M1-M2 polarization axis. Tumor-associated macrophages usually display a M2-like phenotype, associated with pro-tumoral features whereas M1 macrophages exert antitumor functions. Targeting the reprogramming of TAMs toward M1-like macrophages would thus be an efficient way to promote tumor regression. This can be achieved through therapies including chemotherapy, immunotherapy, and radiotherapy (RT). In this review, we first describe how chemo- and immunotherapies can target TAMs and, second, we detail how RT modifies macrophage phenotype and present the molecular pathways that may be involved. The identification of irradiation dose inducing macrophage reprogramming and of the underlying mechanisms could lead to the design of novel therapeutic strategies and improve synergy in combined treatments.
Keywords: chemotherapy; nuclear factor kappa B; polarization immunotherapy; radiotherapy; reactive oxygen species; reprogramming; tumor-associated macrophages.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources