

Human Leukocyte Antigen (HLA) and Immune Regulation: How Do Classical and Non-Classical HLA Alleles Modulate Immune Response to Human Immunodeficiency Virus and Hepatitis C Virus Infections?
- PMID: 28769934
- PMCID: PMC5513977
- DOI: 10.3389/fimmu.2017.00832
Human Leukocyte Antigen (HLA) and Immune Regulation: How Do Classical and Non-Classical HLA Alleles Modulate Immune Response to Human Immunodeficiency Virus and Hepatitis C Virus Infections?
Abstract
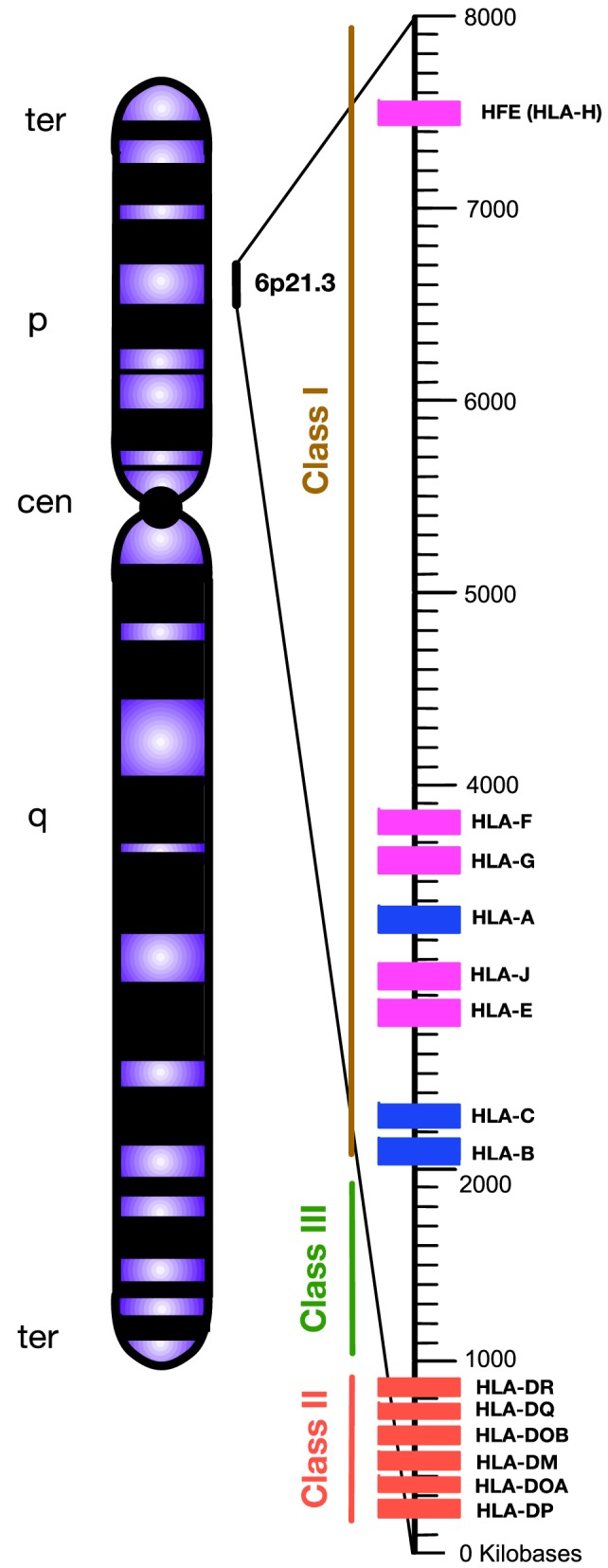
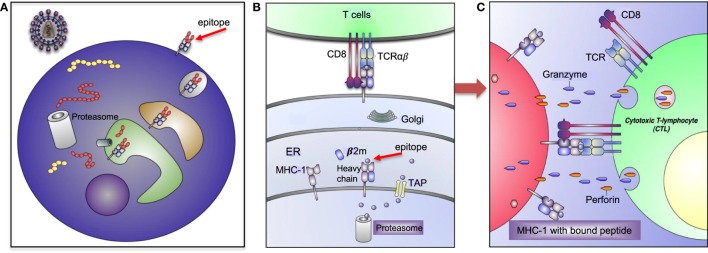
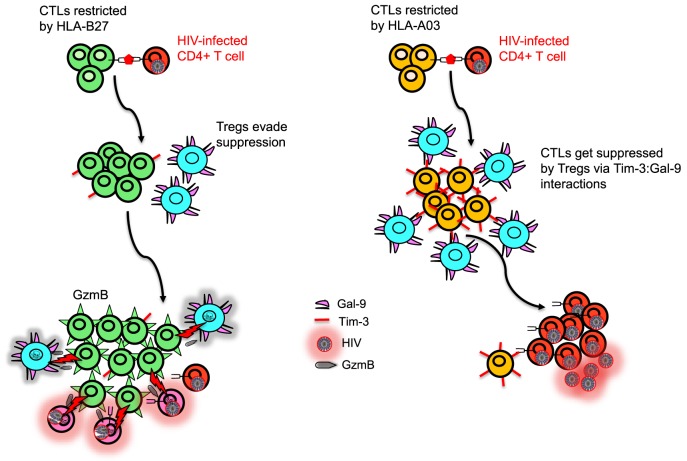
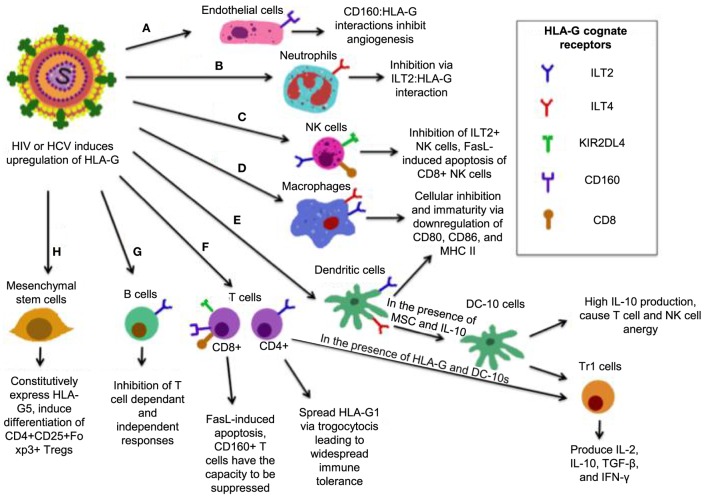
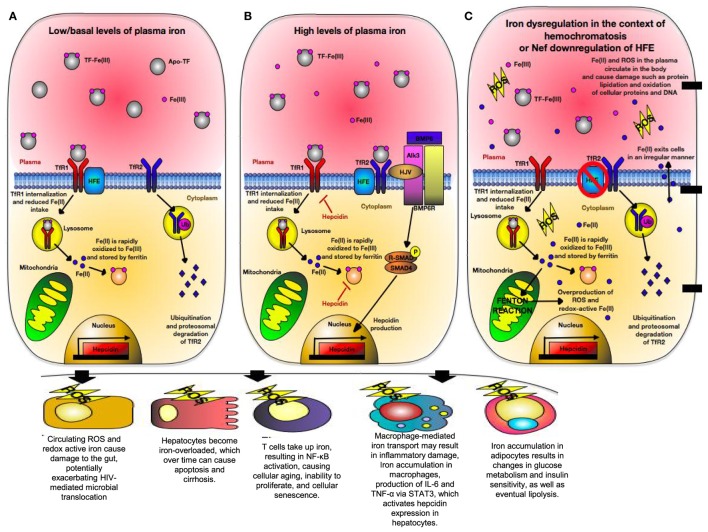
The genetic factors associated with susceptibility or resistance to viral infections are likely to involve a sophisticated array of immune response. These genetic elements may modulate other biological factors that account for significant influence on the gene expression and/or protein function in the host. Among them, the role of the major histocompatibility complex in viral pathogenesis in particular human immunodeficiency virus (HIV) and hepatitis C virus (HCV), is very well documented. We, recently, added a novel insight into the field by identifying the molecular mechanism associated with the protective role of human leukocyte antigen (HLA)-B27/B57 CD8+ T cells in the context of HIV-1 infection and why these alleles act as a double-edged sword protecting against viral infections but predisposing the host to autoimmune diseases. The focus of this review will be reexamining the role of classical and non-classical HLA alleles, including class Ia (HLA-A, -B, -C), class Ib (HLA-E, -F, -G, -H), and class II (HLA-DR, -DQ, -DM, and -DP) in immune regulation and viral pathogenesis (e.g., HIV and HCV). To our knowledge, this is the very first review of its kind to comprehensively analyze the role of these molecules in immune regulation associated with chronic viral infections.
Keywords: classical human leukocyte antigens; hepatitis C virus and immune regulation; human immunodeficiency virus; human leukocyte antigen; major histocompatibility complex; non-classical human leukocyte antigens.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials