

AAA+ Machines of Protein Destruction in Mycobacteria
- PMID: 28770209
- PMCID: PMC5515868
- DOI: 10.3389/fmolb.2017.00049
AAA+ Machines of Protein Destruction in Mycobacteria
Abstract
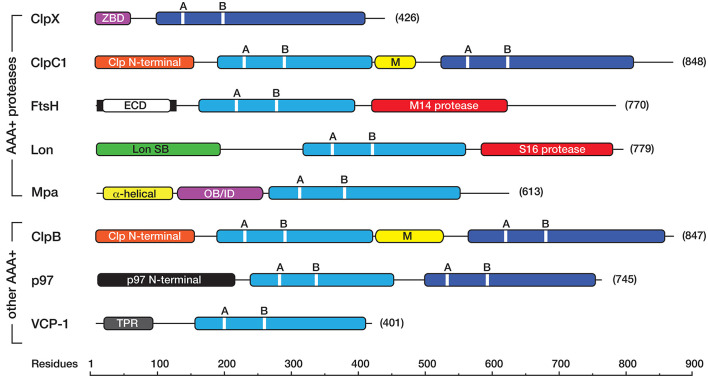
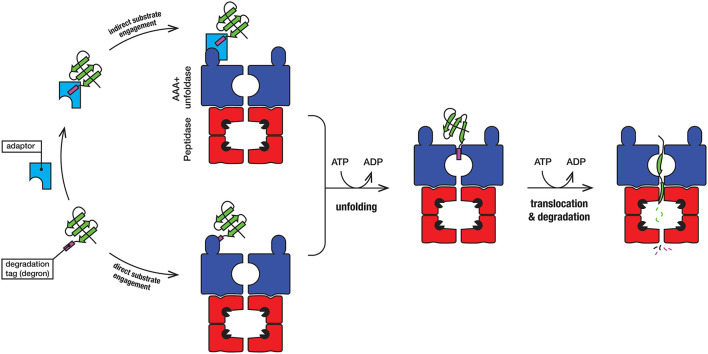
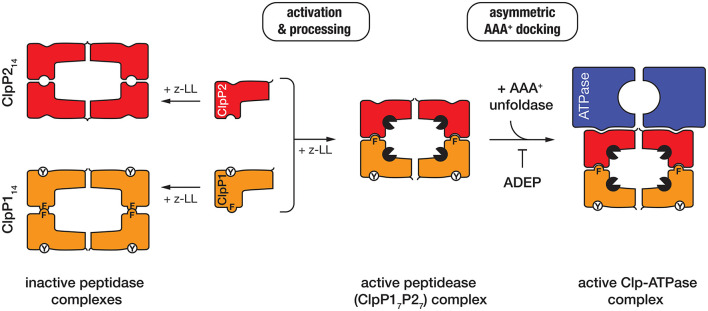
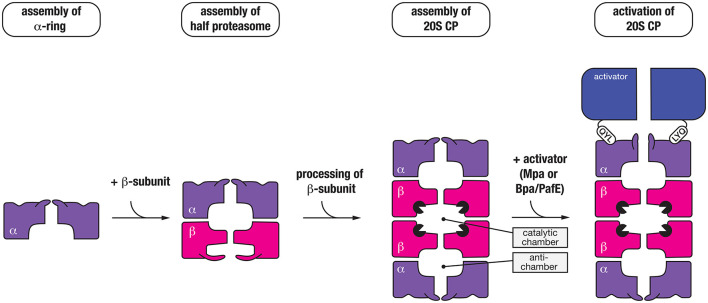
The bacterial cytosol is a complex mixture of macromolecules (proteins, DNA, and RNA), which collectively are responsible for an enormous array of cellular tasks. Proteins are central to most, if not all, of these tasks and as such their maintenance (commonly referred to as protein homeostasis or proteostasis) is vital for cell survival during normal and stressful conditions. The two key aspects of protein homeostasis are, (i) the correct folding and assembly of proteins (coupled with their delivery to the correct cellular location) and (ii) the timely removal of unwanted or damaged proteins from the cell, which are performed by molecular chaperones and proteases, respectively. A major class of proteins that contribute to both of these tasks are the AAA+ (ATPases associated with a variety of cellular activities) protein superfamily. Although much is known about the structure of these machines and how they function in the model Gram-negative bacterium Escherichia coli, we are only just beginning to discover the molecular details of these machines and how they function in mycobacteria. Here we review the different AAA+ machines, that contribute to proteostasis in mycobacteria. Primarily we will focus on the recent advances in the structure and function of AAA+ proteases, the substrates they recognize and the cellular pathways they control. Finally, we will discuss the recent developments related to these machines as novel drug targets.
Keywords: AAA+ protease complexes; Mycobacterium; novel drug targets; proteasome; protein degradation.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The reviewer CE and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
Figures
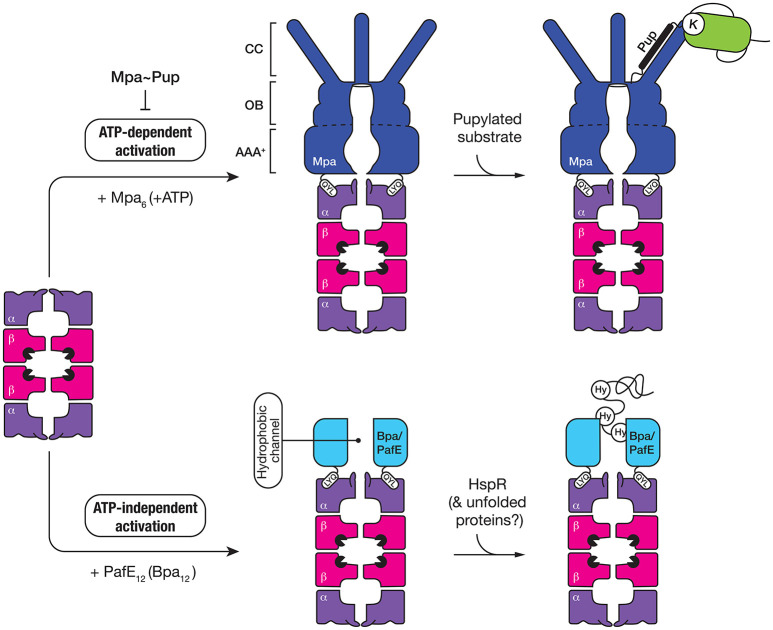
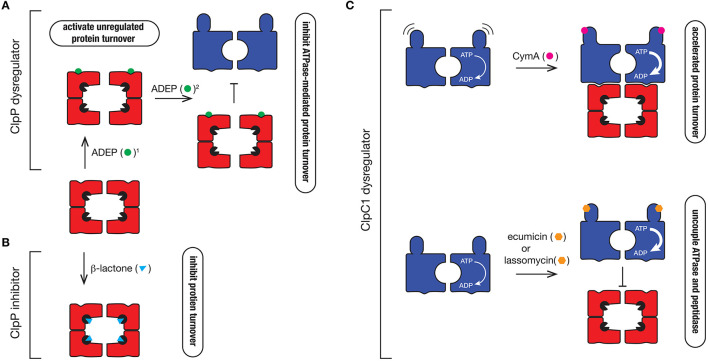
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources