

Diagnosis and treatment of tyrosinemia type I: a US and Canadian consensus group review and recommendations
- PMID: 28771246
- PMCID: PMC5729346
- DOI: 10.1038/gim.2017.101
Diagnosis and treatment of tyrosinemia type I: a US and Canadian consensus group review and recommendations
Abstract
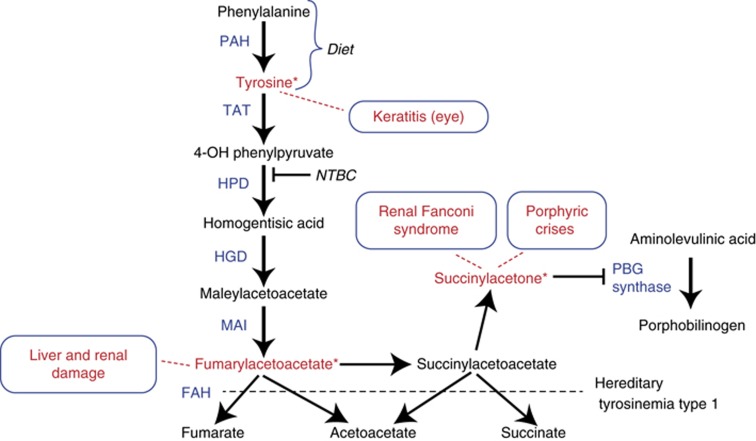
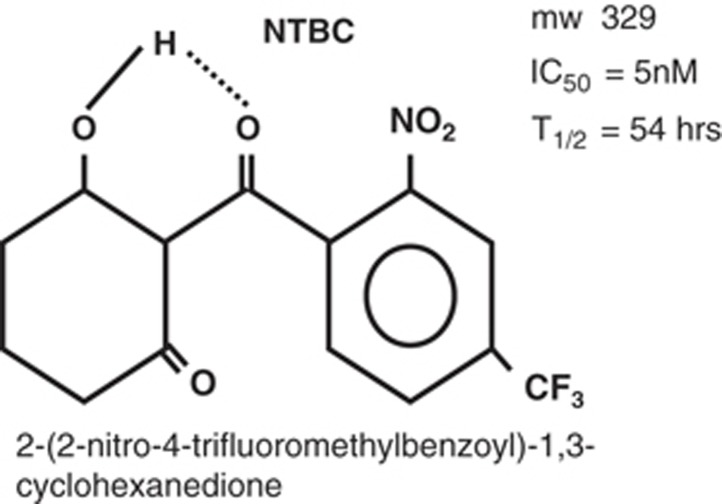
Tyrosinemia type I (hepatorenal tyrosinemia, HT-1) is an autosomal recessive condition resulting in hepatic failure with comorbidities involving the renal and neurologic systems and long term risks for hepatocellular carcinoma. An effective medical treatment with 2-[2-nitro-4-trifluoromethylbenzoyl]-1,3-cyclohexanedione (NTBC) exists but requires early identification of affected children for optimal long-term results. Newborn screening (NBS) utilizing blood succinylacetone as the NBS marker is superior to observing tyrosine levels as a way of identifying neonates with HT-1. If identified early and treated appropriately, the majority of affected infants can remain asymptomatic. A clinical management scheme is needed for infants with HT-1 identified by NBS or clinical symptoms. To this end, a group of 11 clinical practitioners, including eight biochemical genetics physicians, two metabolic dietitian nutritionists, and a clinical psychologist, from the United States and Canada, with experience in providing care for patients with HT-1, initiated an evidence- and consensus-based process to establish uniform recommendations for identification and treatment of HT-1. Recommendations were developed from a literature review, practitioner management survey, and nominal group process involving two face-to-face meetings. There was strong consensus in favor of NBS for HT-1, using blood succinylacetone as a marker, followed by diagnostic confirmation and early treatment with NTBC and diet. Consensus recommendations for both immediate and long-term clinical follow-up of positive diagnoses via both newborn screening and clinical symptomatic presentation are provided.
Conflict of interest statement
All discussions of both the content and writing of the manuscript were performed without the presence or input of any representative of Sobi, Inc. J.M.C., R.S., C.F., M.G.C., M.G., G.M., and C.R.S. have participated as physician consultants/advisers intermittently for Sobi, Inc., the manufacturers of Orfadin (nitisinone) and have received compensation for their activities. The other authors declare no conflict of interest.
Figures
Comment in
-
Heme as an initial treatment for severe decompensation in tyrosinemia type 1.Genet Med. 2020 Feb;22(2):437-438. doi: 10.1038/s41436-019-0658-z. Epub 2019 Oct 1. Genet Med. 2020. PMID: 31570800 No abstract available.
-
Response to Neeleman et al.Genet Med. 2020 Feb;22(2):439-440. doi: 10.1038/s41436-019-0659-y. Epub 2019 Oct 1. Genet Med. 2020. PMID: 31570801 No abstract available.
References
-
- Mitchell GA, Grompe M, Lambert M, Tanguay RM. Hypertyrosinemia. In: Valle D, Beaudet AL, Vogelstein B et al. The Online Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw-Hill, 2014. Available at: http://ommbid.mhmedical.com/content.aspx?bookid=971§ionid=62673883.
-
- Mitchell G, Larochelle J, Lambert M et al. Neurologic crises in hereditary tyrosinemia. N Engl J Med 1990;322:432–437. - PubMed
-
- Castilloux J, Laberge AM, Martin SR, Lallier M, Marchand V. “Silent” tyrosinemia presenting as hepatocellular carcinoma in a 10-year-old girl. J Pediatr Gastroenterol Nutr 2007;44:375–377. - PubMed
-
- Weinberg AG, Mize CE, Worthen HG. The occurrence of hepatoma in the chronic form of hereditary tyrosinemia. J Pediatr 1976;88:434–438. - PubMed
-
- van Spronsen FJ, Thomasse Y, Smit GP et al. Hereditary tyrosinemia type I: a new clinical classification with difference in prognosis on dietary treatment. Hepatology 1994;20:1187–1191. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
