

Assembly and analysis of 100 full MHC haplotypes from the Danish population
- PMID: 28774965
- PMCID: PMC5580718
- DOI: 10.1101/gr.218891.116
Assembly and analysis of 100 full MHC haplotypes from the Danish population
Abstract
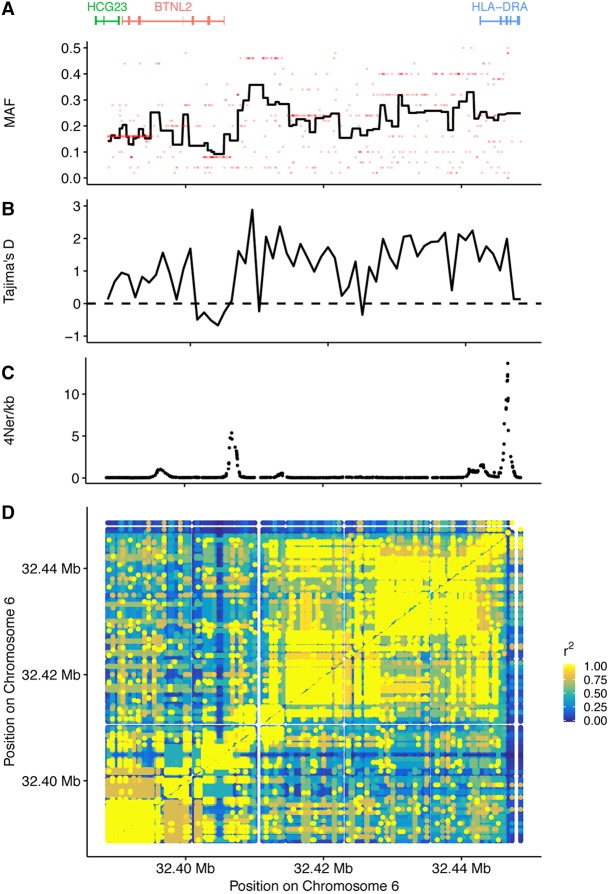
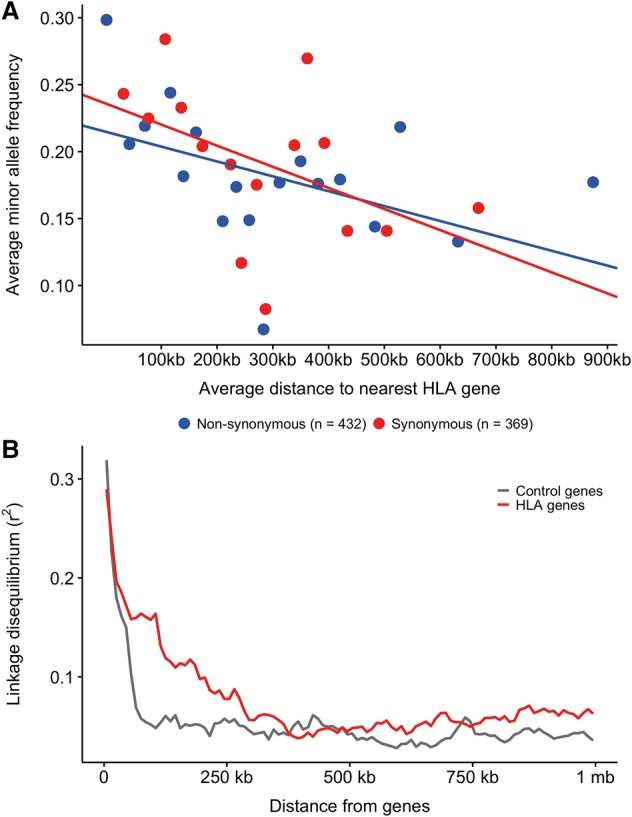
Genes in the major histocompatibility complex (MHC, also known as HLA) play a critical role in the immune response and variation within the extended 4-Mb region shows association with major risks of many diseases. Yet, deciphering the underlying causes of these associations is difficult because the MHC is the most polymorphic region of the genome with a complex linkage disequilibrium structure. Here, we reconstruct full MHC haplotypes from de novo assembled trios without relying on a reference genome and perform evolutionary analyses. We report 100 full MHC haplotypes and call a large set of structural variants in the regions for future use in imputation with GWAS data. We also present the first complete analysis of the recombination landscape in the entire region and show how balancing selection at classical genes have linked effects on the frequency of variants throughout the region.
© 2017 Jensen et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215: 403–410. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous