

Brain-Heart Interaction: Cardiac Complications After Stroke
- PMID: 28775014
- PMCID: PMC5553569
- DOI: 10.1161/CIRCRESAHA.117.311170
Brain-Heart Interaction: Cardiac Complications After Stroke
Abstract
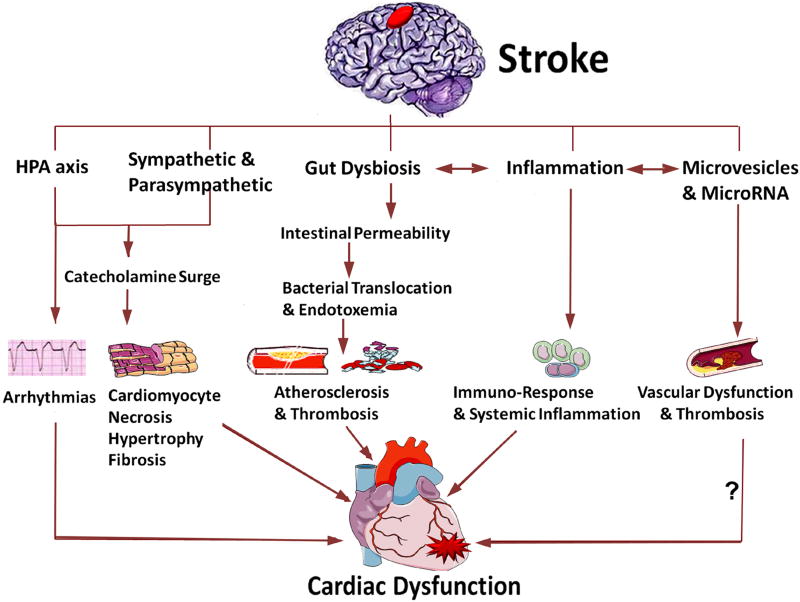
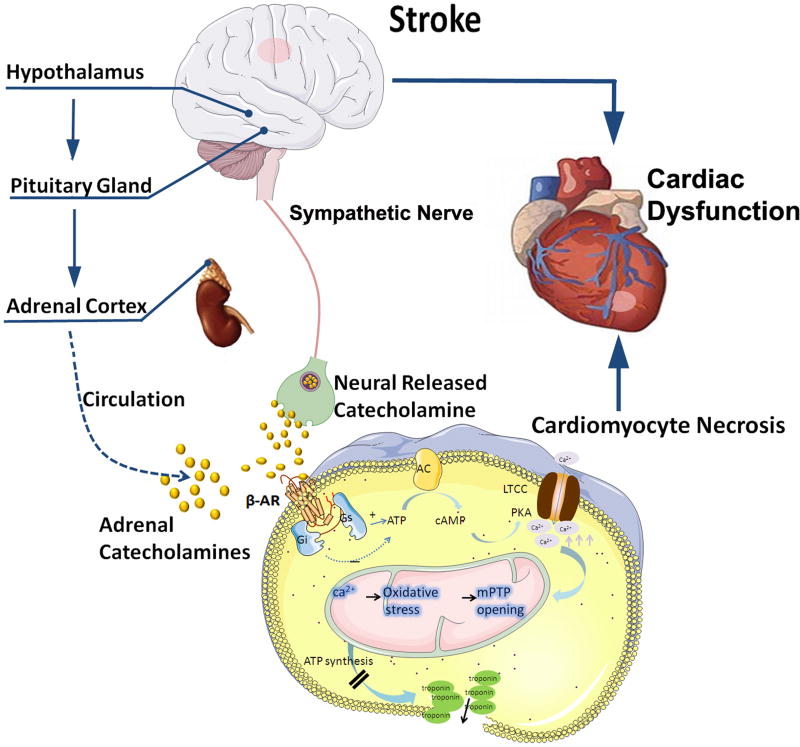
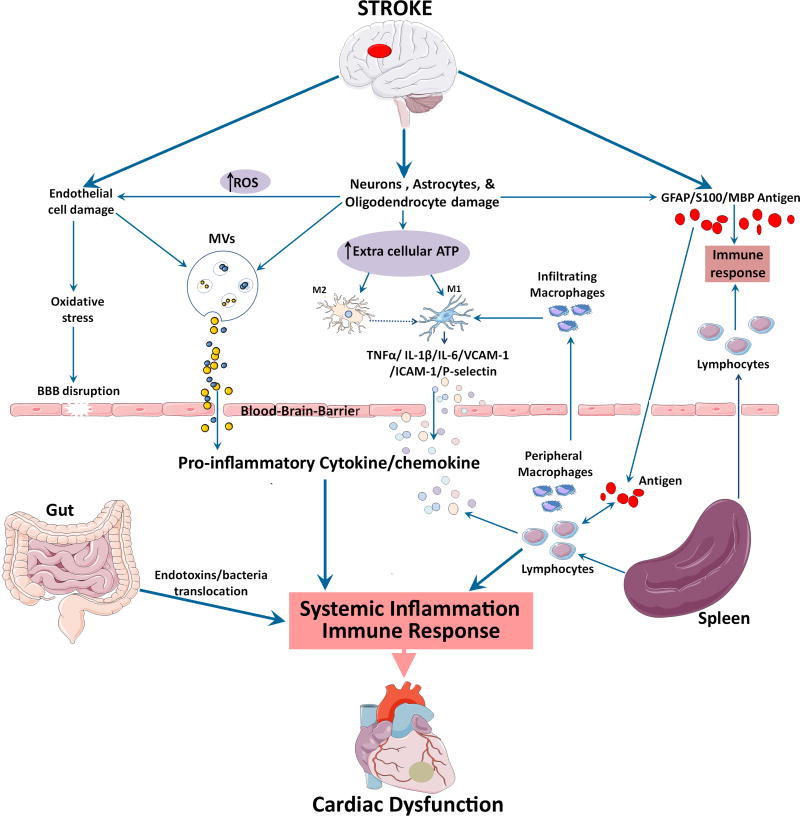
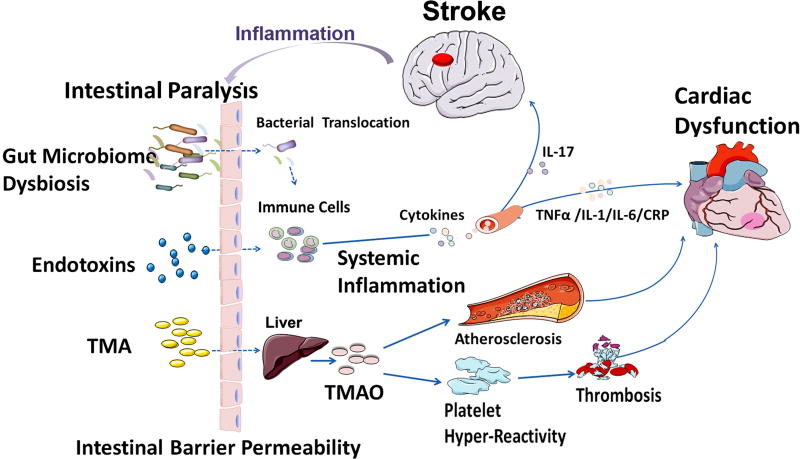
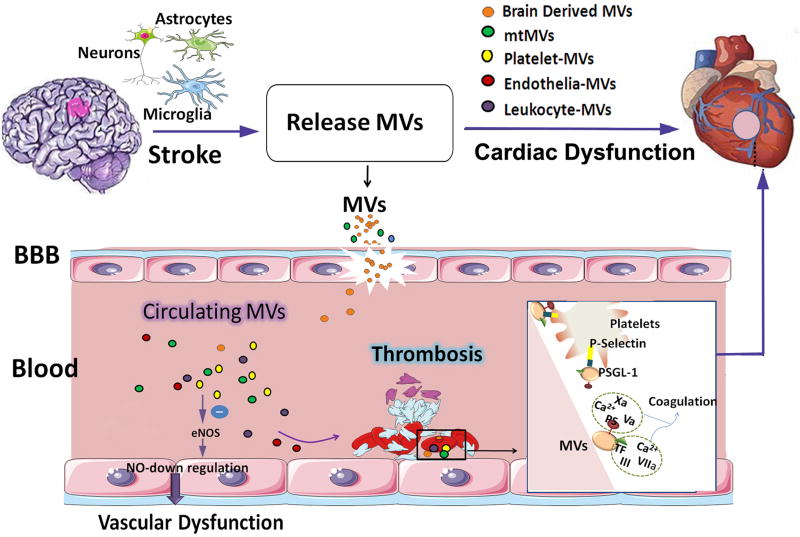
Neurocardiology is an emerging specialty that addresses the interaction between the brain and the heart, that is, the effects of cardiac injury on the brain and the effects of brain injury on the heart. This review article focuses on cardiac dysfunction in the setting of stroke such as ischemic stroke, brain hemorrhage, and subarachnoid hemorrhage. The majority of post-stroke deaths are attributed to neurological damage, and cardiovascular complications are the second leading cause of post-stroke mortality. Accumulating clinical and experimental evidence suggests a causal relationship between brain damage and heart dysfunction. Thus, it is important to determine whether cardiac dysfunction is triggered by stroke, is an unrelated complication, or is the underlying cause of stroke. Stroke-induced cardiac damage may lead to fatality or potentially lifelong cardiac problems (such as heart failure), or to mild and recoverable damage such as neurogenic stress cardiomyopathy and Takotsubo cardiomyopathy. The role of location and lateralization of brain lesions after stroke in brain-heart interaction; clinical biomarkers and manifestations of cardiac complications; and underlying mechanisms of brain-heart interaction after stroke, such as the hypothalamic-pituitary-adrenal axis; catecholamine surge; sympathetic and parasympathetic regulation; microvesicles; microRNAs; gut microbiome, immunoresponse, and systemic inflammation, are discussed.
Keywords: brain injury; brain ischemia; heart failure; inflammation; stroke.
© 2017 American Heart Association, Inc.
Figures
References
-
- Ay H, Koroshetz WJ, Benner T, Vangel MG, Melinosky C, Arsava EM, Ayata C, Zhu M, Schwamm LH, Sorensen AG. Neuroanatomic correlates of stroke-related myocardial injury. Neurology. 2006;66:1325–1329. - PubMed
-
- Oppenheimer SM. Neurogenic cardiac effects of cerebrovascular disease. Current opinion in neurology. 1994;7:20–24. - PubMed
-
- Tokgozoglu SL, Batur MK, Topcuoglu MA, Saribas O, Kes S, Oto A. Effects of stroke localization on cardiac autonomic balance and sudden death. Stroke; a journal of cerebral circulation. 1999;30:1307–1311. - PubMed
-
- Byer E, Ashman R, Toth LA. Electrocardiograms with large, upright t waves and long q-t intervals. American heart journal. 1947;33:796–806. - PubMed
-
- Samuels MA. The brain-heart connection. Circulation. 2007;116:77–84. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
