

Cytokines and Chemokines in Cerebral Malaria Pathogenesis
- PMID: 28775960
- PMCID: PMC5517394
- DOI: 10.3389/fcimb.2017.00324
Cytokines and Chemokines in Cerebral Malaria Pathogenesis
Abstract
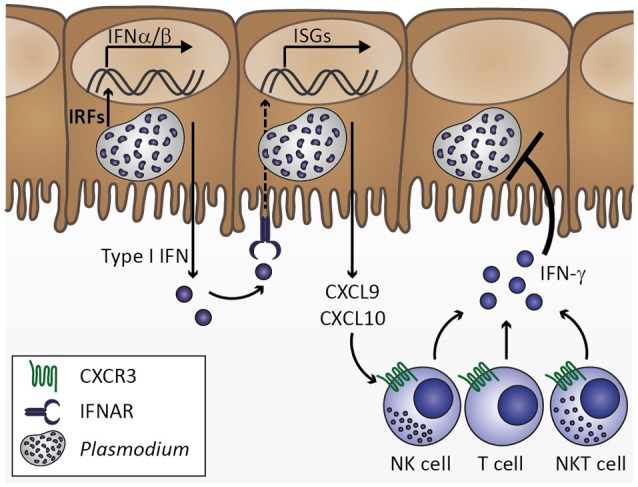
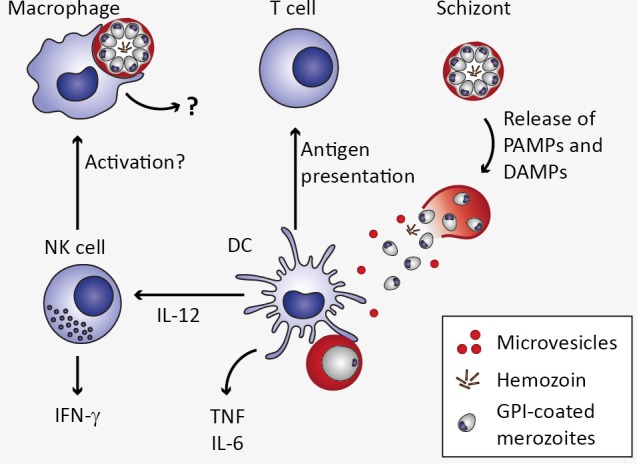
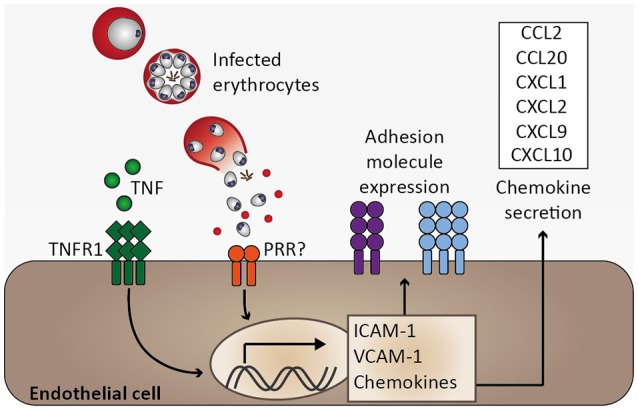
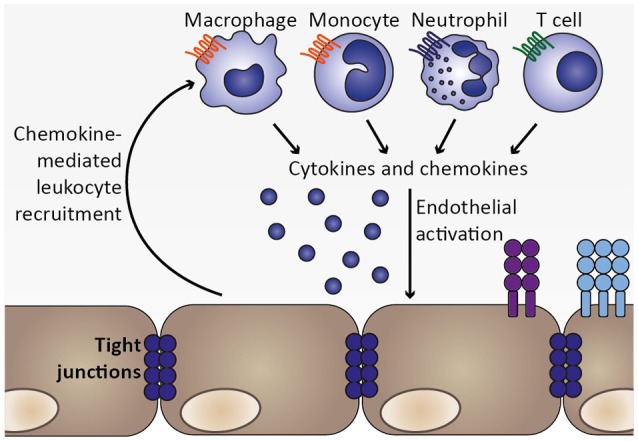
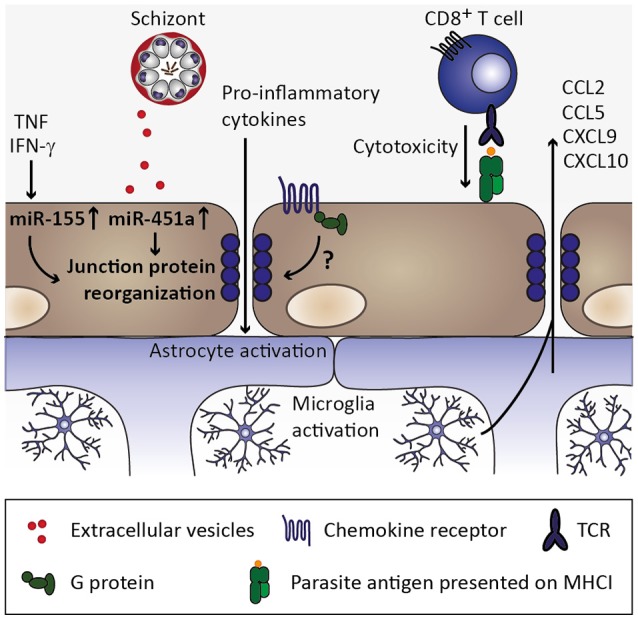
Cerebral malaria is among the major causes of malaria-associated mortality and effective adjunctive therapeutic strategies are currently lacking. Central pathophysiological processes involved in the development of cerebral malaria include an imbalance of pro- and anti-inflammatory responses to Plasmodium infection, endothelial cell activation, and loss of blood-brain barrier integrity. However, the sequence of events, which initiates these pathophysiological processes as well as the contribution of their complex interplay to the development of cerebral malaria remain incompletely understood. Several cytokines and chemokines have repeatedly been associated with cerebral malaria severity. Increased levels of these inflammatory mediators could account for the sequestration of leukocytes in the cerebral microvasculature present during cerebral malaria, thereby contributing to an amplification of local inflammation and promoting cerebral malaria pathogenesis. Herein, we highlight the current knowledge on the contribution of cytokines and chemokines to the pathogenesis of cerebral malaria with particular emphasis on their roles in endothelial activation and leukocyte recruitment, as well as their implication in the progression to blood-brain barrier permeability and neuroinflammation, in both human cerebral malaria and in the murine experimental cerebral malaria model. A better molecular understanding of these processes could provide the basis for evidence-based development of adjunct therapies and the definition of diagnostic markers of disease progression.
Keywords: Plasmodium; blood-brain barrier; cerebral malaria; chemokines; cytokines; endothelial activation; malaria; neuroinflammation.
Figures

References
-
- Adams Y., Kuhnrae P., Higgins M. K., Ghumra A., Rowe J. A. (2014). Rosetting Plasmodium falciparum-infected erythrocytes bind to human brain microvascular endothelial cells in vitro, demonstrating a dual adhesion phenotype mediated by distinct P. falciparum erythrocyte membrane protein 1 domains. Infect. Immun. 82, 949–959. 10.1128/IAI.01233-13 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources