

KSHV and the Role of Notch Receptor Dysregulation in Disease Progression
- PMID: 28777778
- PMCID: PMC5617991
- DOI: 10.3390/pathogens6030034
KSHV and the Role of Notch Receptor Dysregulation in Disease Progression
Abstract
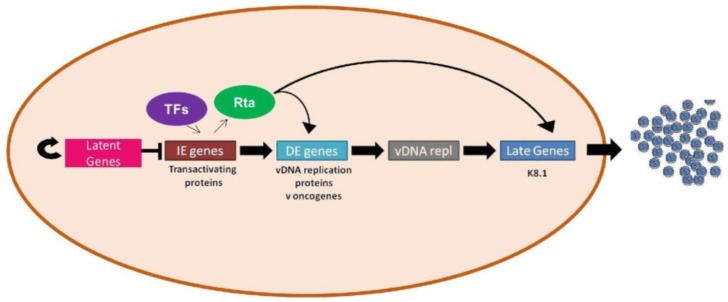
Kaposi's sarcoma-associated herpesvirus (KSHV) is the causative agent of two human cancers, Kaposi's Sarcoma (KS) and primary effusion lymphoma (PEL), and a lymphoproliferation, Multicentric Castleman's Disease (MCD). Progression to tumor development in KS is dependent upon the reactivation of the virus from its latent state. We, and others, have shown that the Replication and transcriptional activator (Rta) protein is the only viral gene product that is necessary and sufficient for viral reactivation. To induce the reactivation and transcription of viral genes, Rta forms a complex with the cellular DNA binding component of the canonical Notch signaling pathway, recombination signal binding protein for Jk (RBP-Jk). Formation of this Rta:RBP-Jk complex is necessary for viral reactivation to occur. Expression of activated Notch has been shown to be dysregulated in KSHV infected cells and to be necessary for cell growth and disease progression. Studies into the involvement of activated Notch in viral reactivation have yielded varied results. In this paper, we review the current literature regarding Notch dysregulation by KSHV and its role in viral infection and cellular pathogenesis.
Keywords: HHV8; KSHV; RBP-Jk; Rta; kaposi’s sarcoma; multicentric castleman’s disease; notch; primary effusion lymphoma.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
The cellular Notch1 protein promotes KSHV reactivation in an Rta-dependent manner.J Virol. 2024 Aug 20;98(8):e0078824. doi: 10.1128/jvi.00788-24. Epub 2024 Jul 8. J Virol. 2024. PMID: 38975769 Free PMC article.
-
Convergence of Kaposi's sarcoma-associated herpesvirus reactivation with Epstein-Barr virus latency and cellular growth mediated by the notch signaling pathway in coinfected cells.J Virol. 2010 Oct;84(20):10488-500. doi: 10.1128/JVI.00894-10. Epub 2010 Aug 4. J Virol. 2010. PMID: 20686042 Free PMC article.
-
Genome-Wide Identification of Direct RTA Targets Reveals Key Host Factors for Kaposi's Sarcoma-Associated Herpesvirus Lytic Reactivation.J Virol. 2019 Feb 19;93(5):e01978-18. doi: 10.1128/JVI.01978-18. Print 2019 Mar 1. J Virol. 2019. PMID: 30541837 Free PMC article.
-
[Replication Machinery of Kaposi's Sarcoma-associated Herpesvirus and Drug Discovery Research].Yakugaku Zasshi. 2019;139(1):69-73. doi: 10.1248/yakushi.18-00164-2. Yakugaku Zasshi. 2019. PMID: 30606932 Review. Japanese.
-
The pleiotropic effects of Kaposi's sarcoma herpesvirus.J Pathol. 2006 Jan;208(2):187-98. doi: 10.1002/path.1904. J Pathol. 2006. PMID: 16362980 Review.
Cited by
-
Transcriptional and post-transcriptional regulation of viral gene expression in the gamma-herpesvirus Kaposi's sarcoma-associated herpesvirus.Curr Clin Microbiol Rep. 2018 Dec;5(4):219-228. doi: 10.1007/s40588-018-0102-1. Epub 2018 Aug 3. Curr Clin Microbiol Rep. 2018. PMID: 30854283 Free PMC article.
-
Inhibition of MEK-ERK pathway enhances oncolytic vaccinia virus replication in doxorubicin-resistant ovarian cancer.Mol Ther Oncolytics. 2022 Apr 18;25:211-224. doi: 10.1016/j.omto.2022.04.006. eCollection 2022 Jun 16. Mol Ther Oncolytics. 2022. PMID: 35592390 Free PMC article.
-
Primary effusion lymphoma: current perspectives.Onco Targets Ther. 2018 Jun 28;11:3747-3754. doi: 10.2147/OTT.S167392. eCollection 2018. Onco Targets Ther. 2018. PMID: 29988764 Free PMC article. Review.
-
Whole brain radiation therapy resulting in radionecrosis: a possible link with radiosensitising chemoimmunotherapy.BMJ Case Rep. 2023 Nov 28;16(11):e256758. doi: 10.1136/bcr-2023-256758. BMJ Case Rep. 2023. PMID: 38016763
-
LncRNA-HANR exacerbates malignant behaviors of cholangiocarcinoma cells through activating Notch pathway.Heliyon. 2023 Nov 11;9(12):e22087. doi: 10.1016/j.heliyon.2023.e22087. eCollection 2023 Dec. Heliyon. 2023. PMID: 38076116 Free PMC article.
References
-
- Soulier J., Grollet L., Oksenhendler E., Cacoub P., Cazals-Hatem D., Babinet P., d’Agay M., Clauvel J., Raphael M., Degos L., et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood. 1995;86:1276–1280. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
