

Heterozygous De Novo UBTF Gain-of-Function Variant Is Associated with Neurodegeneration in Childhood
- PMID: 28777933
- PMCID: PMC5544390
- DOI: 10.1016/j.ajhg.2017.07.002
Heterozygous De Novo UBTF Gain-of-Function Variant Is Associated with Neurodegeneration in Childhood
Abstract
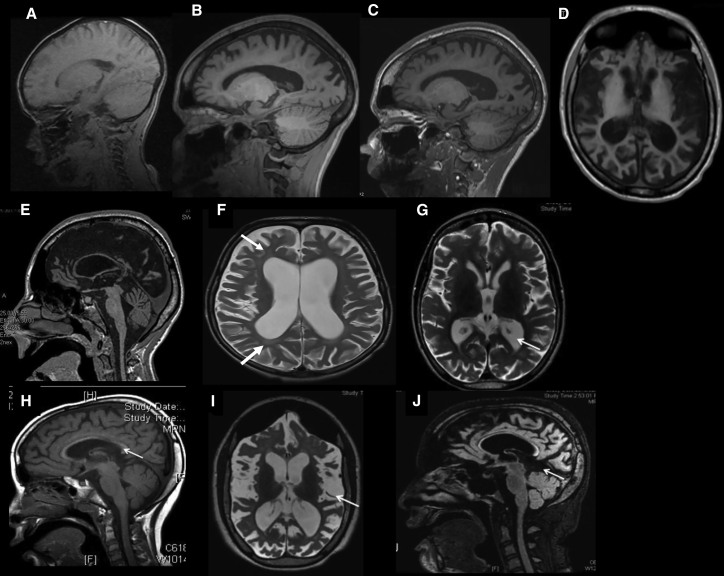
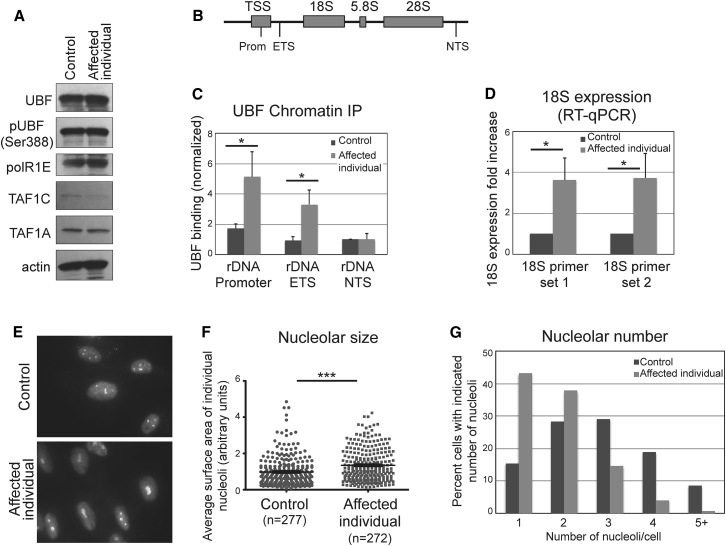
Ribosomal RNA (rRNA) is transcribed from rDNA by RNA polymerase I (Pol I) to produce the 45S precursor of the 28S, 5.8S, and 18S rRNA components of the ribosome. Two transcription factors have been defined for Pol I in mammals, the selectivity factor SL1, and the upstream binding transcription factor (UBF), which interacts with the upstream control element to facilitate the assembly of the transcription initiation complex including SL1 and Pol I. In seven unrelated affected individuals, all suffering from developmental regression starting at 2.5-7 years, we identified a heterozygous variant, c.628G>A in UBTF, encoding p.Glu210Lys in UBF, which occurred de novo in all cases. While the levels of UBF, Ser388 phosphorylated UBF, and other Pol I-related components (POLR1E, TAF1A, and TAF1C) remained unchanged in cells of an affected individual, the variant conferred gain of function to UBF, manifesting by markedly increased UBF binding to the rDNA promoter and to the 5'- external transcribed spacer. This was associated with significantly increased 18S expression, and enlarged nucleoli which were reduced in number per cell. The data link neurodegeneration in childhood with altered rDNA chromatin status and rRNA metabolism.
Copyright © 2017 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Crumrine P.K. Degenerative disorders of the central nervous system. Pediatr. Rev. 2001;22:370–379. - PubMed
-
- Ta-Shma A., Zhang K., Salimova E., Zernecke A., Sieiro-Mosti D., Stegner D., Furtado M., Shaag A., Perles Z., Nieswandt B. Congenital valvular defects associated with deleterious mutations in the PLD1 gene. J. Med. Genet. 2016 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
