

Mutations in TRAPPC12 Manifest in Progressive Childhood Encephalopathy and Golgi Dysfunction
- PMID: 28777934
- PMCID: PMC5544387
- DOI: 10.1016/j.ajhg.2017.07.006
Mutations in TRAPPC12 Manifest in Progressive Childhood Encephalopathy and Golgi Dysfunction
Abstract
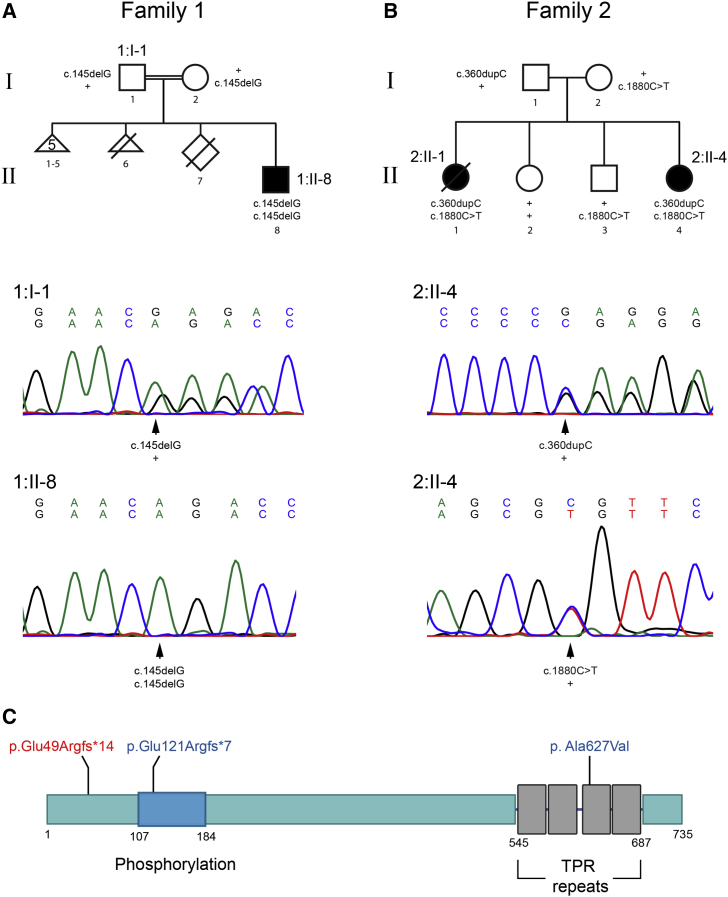
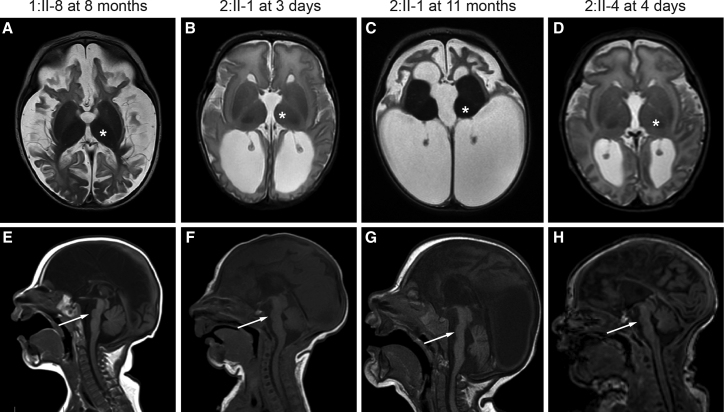
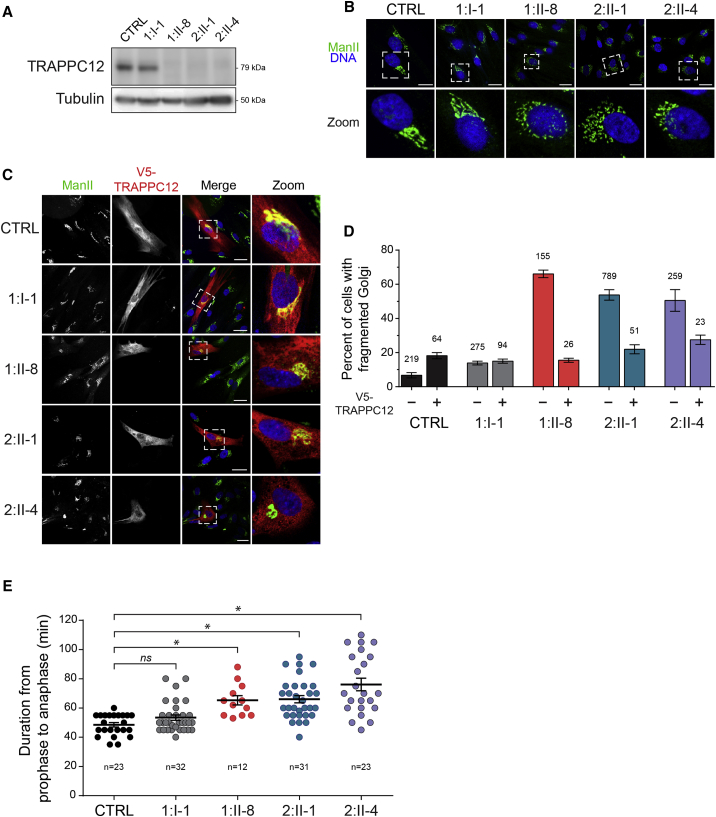
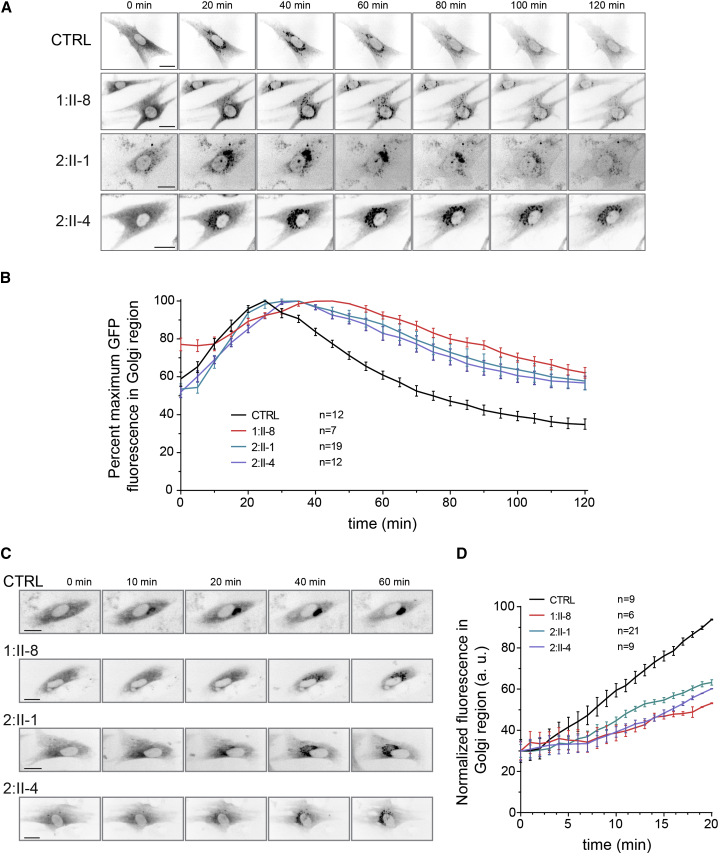
Progressive childhood encephalopathy is an etiologically heterogeneous condition characterized by progressive central nervous system dysfunction in association with a broad range of morbidity and mortality. The causes of encephalopathy can be either non-genetic or genetic. Identifying the genetic causes and dissecting the underlying mechanisms are critical to understanding brain development and improving treatments. Here, we report that variants in TRAPPC12 result in progressive childhood encephalopathy. Three individuals from two unrelated families have either a homozygous deleterious variant (c.145delG [p.Glu49Argfs∗14]) or compound-heterozygous variants (c.360dupC [p.Glu121Argfs∗7] and c.1880C>T [p. Ala627Val]). The clinical phenotypes of the three individuals are strikingly similar: severe disability, microcephaly, hearing loss, spasticity, and characteristic brain imaging findings. Fibroblasts derived from all three individuals showed a fragmented Golgi that could be rescued by expression of wild-type TRAPPC12. Protein transport from the endoplasmic reticulum to and through the Golgi was delayed. TRAPPC12 is a member of the TRAPP protein complex, which functions in membrane trafficking. Variants in several other genes encoding members of the TRAPP complex have been associated with overlapping clinical presentations, indicating shared and distinct functions for each complex member. Detailed understanding of the TRAPP-opathies will illuminate the role of membrane protein transport in human disease.
Keywords: Golgi; TRAPP; TRAPPC12; brain atrophy; encephalopathy; potocerebellar hypoplasia.
Copyright © 2017 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Armstrong L., Biancheri R., Shyr C., Rossi A., Sinclair G., Ross C.J., Tarailo-Graovac M., Wasserman W.W., van Karnebeek C.D. AIMP1 deficiency presents as a cortical neurodegenerative disease with infantile onset. Neurogenetics. 2014;15:157–159. - PubMed
-
- Chong J.X., Caputo V., Phelps I.G., Stella L., Worgan L., Dempsey J.C., Nguyen A., Leuzzi V., Webster R., Pizzuti A., University of Washington Center for Mendelian Genomics Recessive Inactivating Mutations in TBCK, Encoding a Rab GTPase-Activating Protein, Cause Severe Infantile Syndromic Encephalopathy. Am. J. Hum. Genet. 2016;98:772–781. - PMC - PubMed
-
- Dimassi S., Labalme A., Ville D., Calender A., Mignot C., Boutry-Kryza N., de Bellescize J., Rivier-Ringenbach C., Bourel-Ponchel E., Cheillan D. Whole-exome sequencing improves the diagnosis yield in sporadic infantile spasm syndrome. Clin. Genet. 2016;89:198–204. - PubMed
-
- Kurata H., Terashima H., Nakashima M., Okazaki T., Matsumura W., Ohno K., Saito Y., Maegaki Y., Kubota M., Nanba E. Characterization of SPATA5-related encephalopathy in early childhood. Clin. Genet. 2016;90:437–444. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
