

De Novo Mutations in YWHAG Cause Early-Onset Epilepsy
- PMID: 28777935
- PMCID: PMC5544417
- DOI: 10.1016/j.ajhg.2017.07.004
De Novo Mutations in YWHAG Cause Early-Onset Epilepsy
Abstract
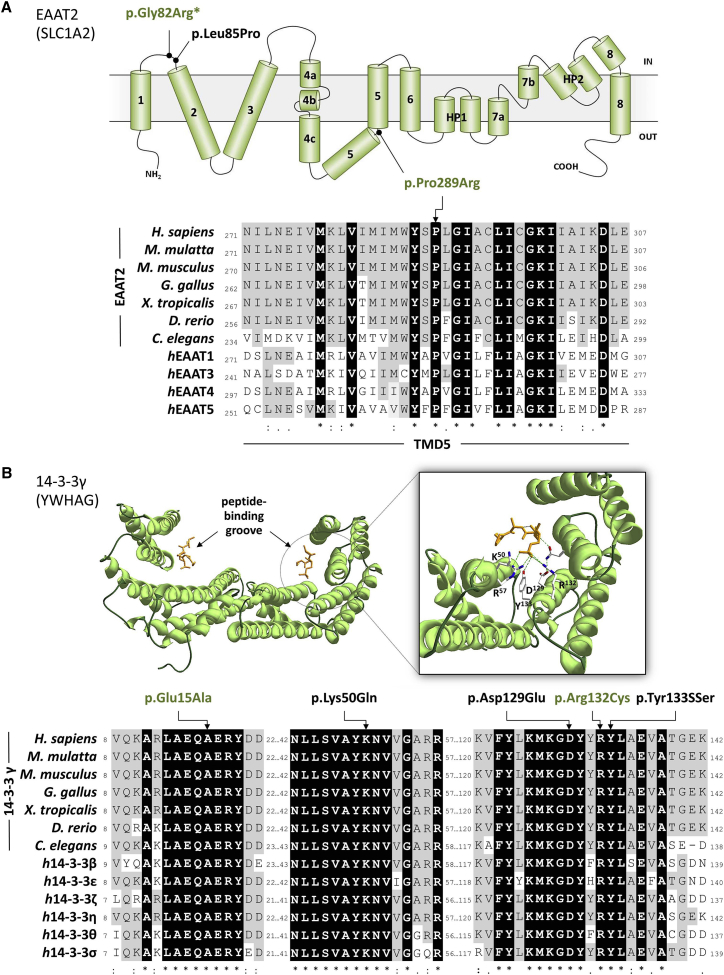
Massively parallel sequencing has revealed many de novo mutations in the etiology of developmental and epileptic encephalopathies (EEs), highlighting their genetic heterogeneity. Additional candidate genes have been prioritized in silico by their co-expression in the brain. Here, we evaluate rare coding variability in 20 candidates nominated with the use of a reference gene set of 51 established EE-associated genes. Variants within the 20 candidate genes were extracted from exome-sequencing data of 42 subjects with EE and no previous genetic diagnosis. We identified 7 rare non-synonymous variants in 7 of 20 genes and performed Sanger sequence validation in affected probands and parental samples. De novo variants were found only in SLC1A2 (aka EAAT2 or GLT1) (c.244G>A [p.Gly82Arg]) and YWHAG (aka 14-3-3γ) (c.394C>T [p.Arg132Cys]), highlighting the potential cause of EE in 5% (2/42) of subjects. Seven additional subjects with de novo variants in SLC1A2 (n = 1) and YWHAG (n = 6) were subsequently identified through online tools. We identified a highly significant enrichment of de novo variants in YWHAG, establishing their role in early-onset epilepsy, and we provide additional support for the prior assignment of SLC1A2. Hence, in silico modeling of brain co-expression is an efficient method for nominating EE-associated genes to further elucidate the disorder's etiology and genotype-phenotype correlations.
Keywords: SLC1A2; YWHAG; de novo variants; epileptic encephalopathy; whole-exome sequencing.
Copyright © 2017 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
