

Genetic PrP Prion Diseases
- PMID: 28778873
- PMCID: PMC5932589
- DOI: 10.1101/cshperspect.a033134
Genetic PrP Prion Diseases
Abstract
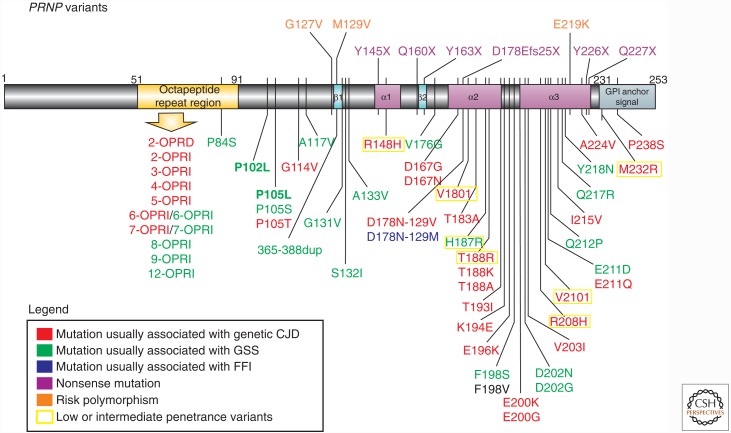
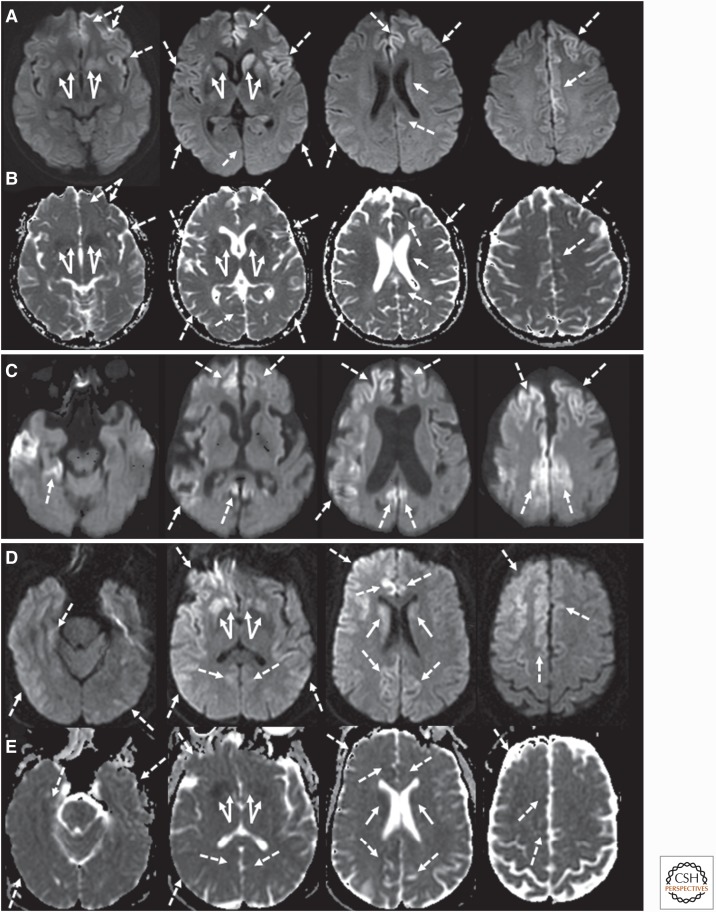
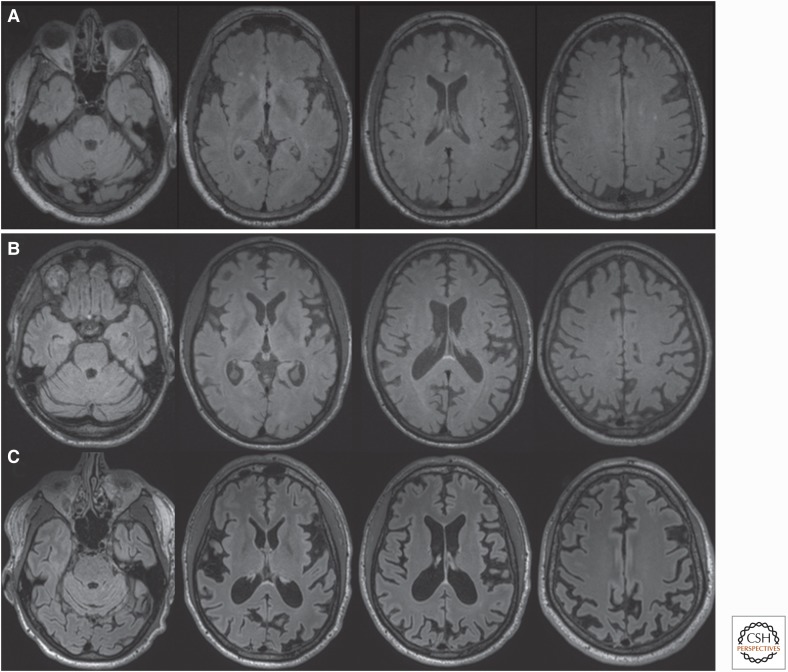
Genetic prion diseases (gPrDs) caused by mutations in the prion protein gene (PRNP) have been classified as genetic Creutzfeldt-Jakob disease, Gerstmann-Sträussler-Scheinker disease, or fatal familial insomnia. Mutations in PRNP can be missense, nonsense, and/or octapeptide repeat insertions or, possibly, deletions. These mutations can produce diverse clinical features. They may also show varying ancillary testing results and neuropathological findings. Although the majority of gPrDs have a rapid progression with a short survival time of a few months, many also present as ataxic or parkinsonian disorders, which have a slower decline over a few to several years. A few very rare mutations manifest as neuropsychiatric disorders, with systemic symptoms that include gastrointestinal disorders and neuropathy; these forms can progress over years to decades. In this review, we classify gPrDs as rapid, slow, or mixed types based on their typical rate of progression and duration, and we review the broad spectrum of phenotypes manifested by these diseases.
Copyright © 2018 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures
References
-
- Alzualde A, Indakoetxea B, Ferrer I, Moreno F, Barandiaran M, Gorostidi A, Estanga A, Ruiz I, Calero M, van Leeuwen FW, et al. 2010. A novel PRNP Y218N mutation in Gerstmann–Sträussler–Scheinker disease with neurofibrillary degeneration. J Neuropathol Exp Neurol 69: 789–800. - PubMed
-
- Beck JA, Mead S, Campbell TA, Dickinson A, Wientjens DP, Croes EA, Van Duijn CM, Collinge J. 2001. Two-octapeptide repeat deletion of prion protein associated with rapidly progressive dementia. Neurology 57: 354–356. - PubMed
-
- Beck JA, Poulter M, Campbell TA, Adamson G, Uphill JB, Guerreiro R, Jackson GS, Stevens JC, Manji H, Collinge J, et al. 2010. PRNP allelic series from 19 years of prion protein gene sequencing at the MRC Prion Unit. Hum Mutat 31: E1551–E1563. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials