

Pyruvate kinase M knockdown-induced signaling via AMP-activated protein kinase promotes mitochondrial biogenesis, autophagy, and cancer cell survival
- PMID: 28778925
- PMCID: PMC5602412
- DOI: 10.1074/jbc.M117.791343
Pyruvate kinase M knockdown-induced signaling via AMP-activated protein kinase promotes mitochondrial biogenesis, autophagy, and cancer cell survival
Abstract
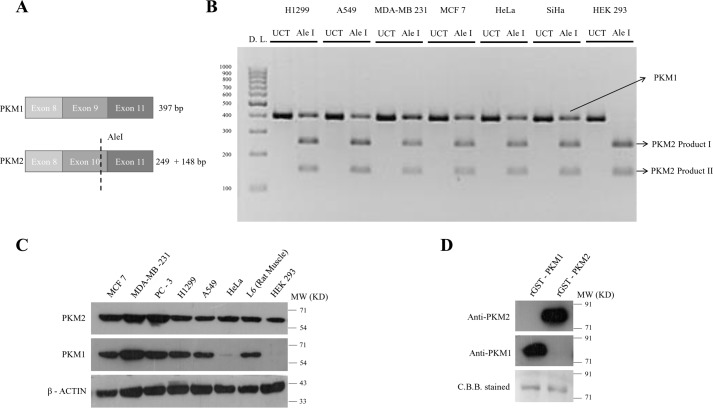
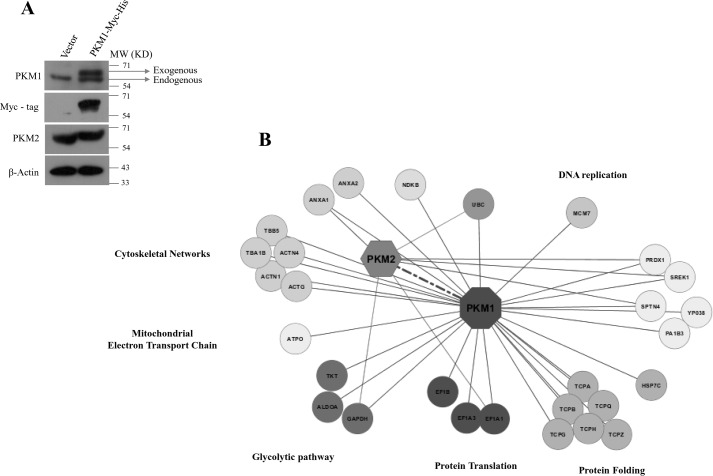
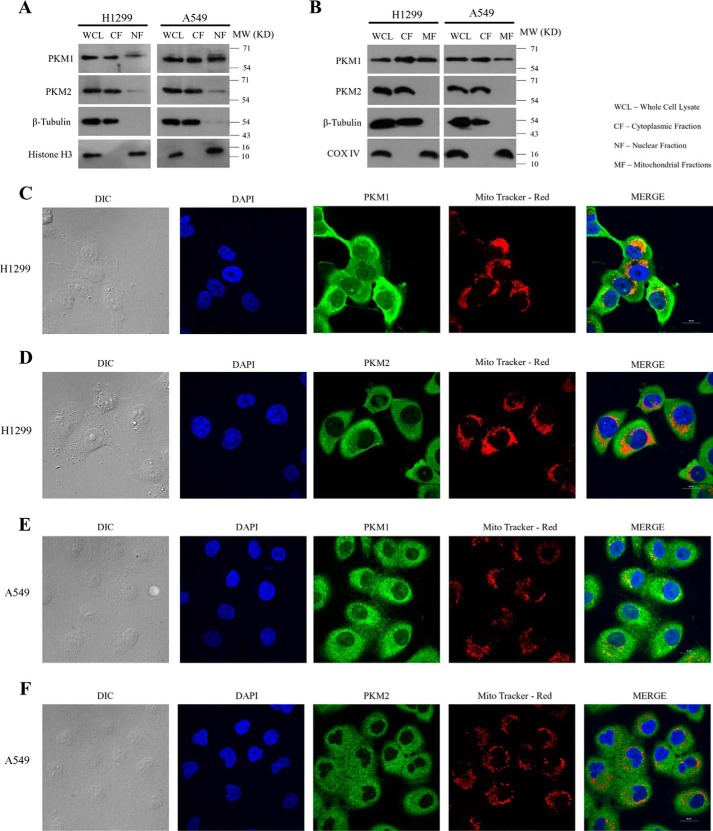
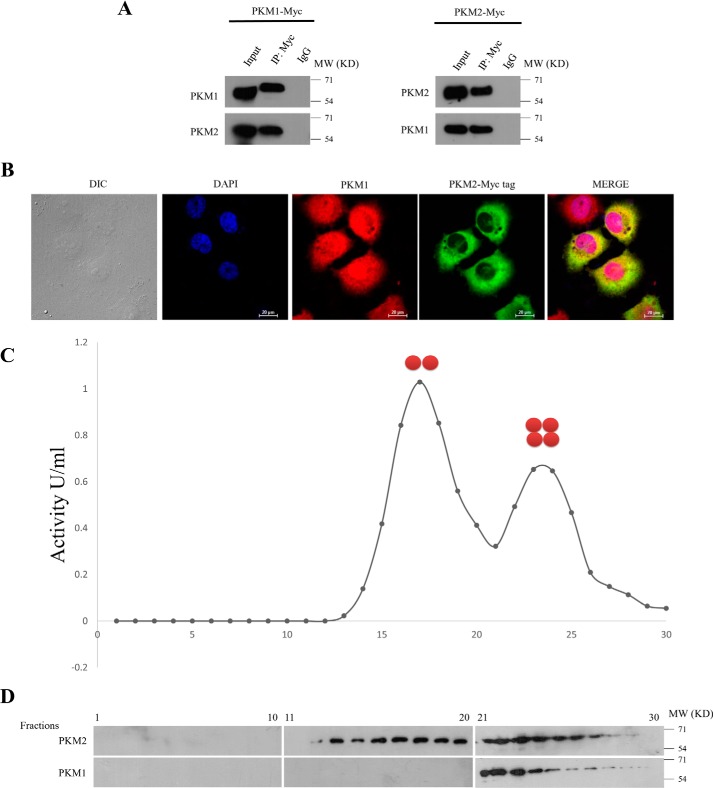
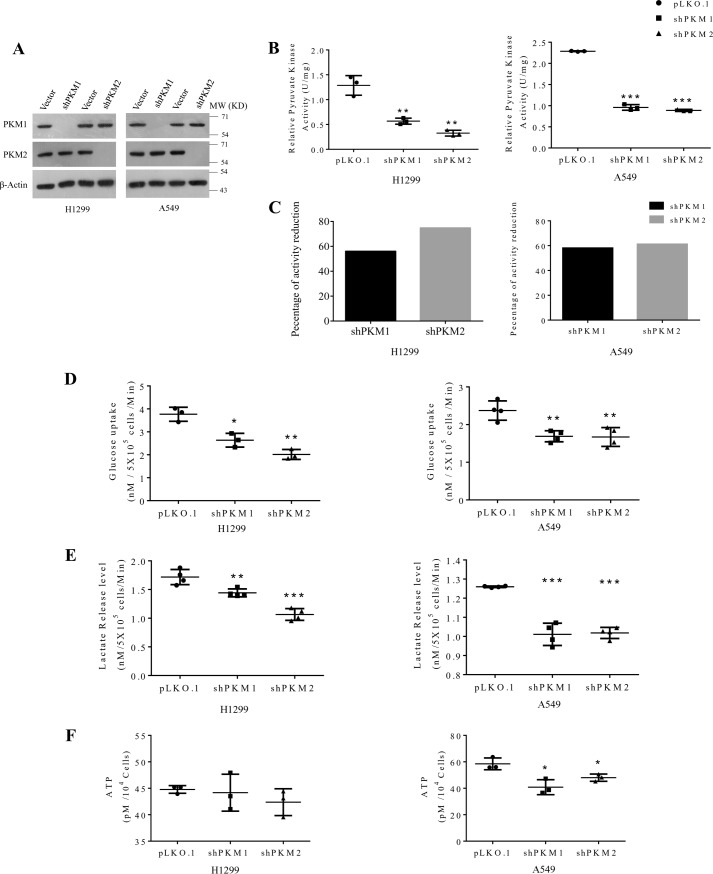
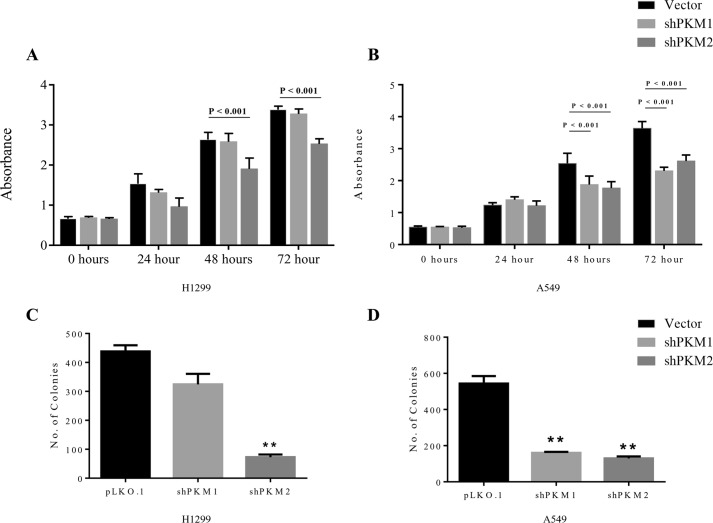
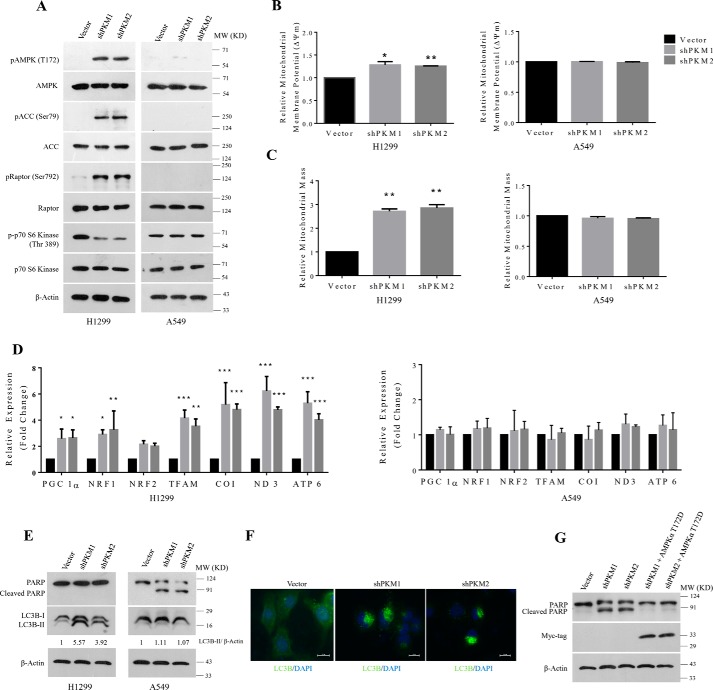
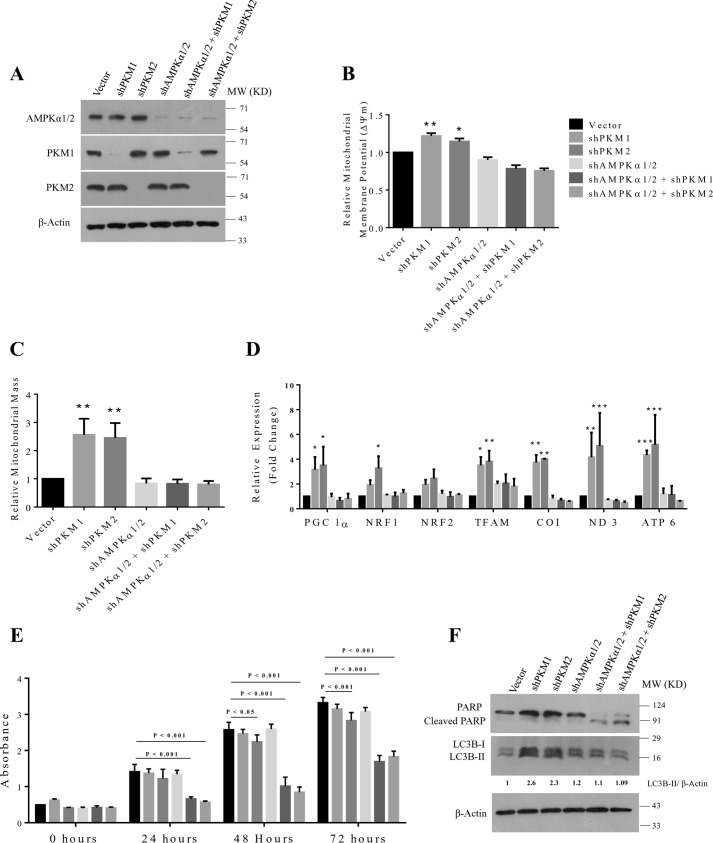
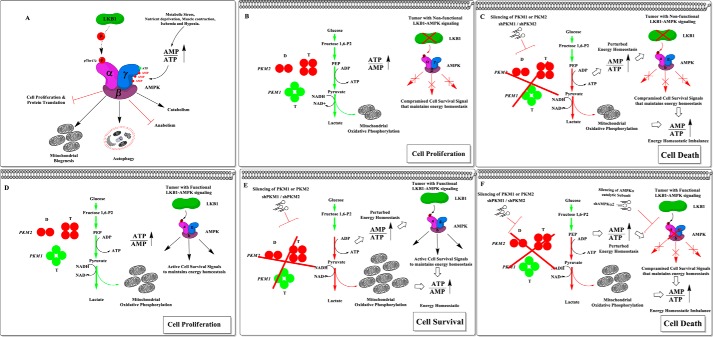
Preferential expression of the low-activity (dimeric) M2 isoform of pyruvate kinase (PK) over its constitutively active splice variant M1 isoform is considered critical for aerobic glycolysis in cancer cells. However, our results reported here indicate co-expression of PKM1 and PKM2 and their possible physical interaction in cancer cells. We show that knockdown of either PKM1 or PKM2 differentially affects net PK activity, viability, and cellular ATP levels of the lung carcinoma cell lines H1299 and A549. The stable knockdown of PK isoforms in A549 cells significantly reduced the cellular ATP level, whereas in H1299 cells the level of ATP was unaltered. Interestingly, the PKM1/2 knockdown in H1299 cells activated AMP-activated protein kinase (AMPK) signaling and stimulated mitochondrial biogenesis and autophagy to maintain energy homeostasis. In contrast, knocking down either of the PKM isoforms in A549 cells lacking LKB1, a serine/threonine protein kinase upstream of AMPK, failed to activate AMPK and sustain energy homeostasis and resulted in apoptosis. Moreover, in a similar genetic background of silenced PKM1 or PKM2, the knocking down of AMPKα1/2 catalytic subunit in H1299 cells induced apoptosis. Our findings help explain why previous targeting of PKM2 in cancer cells to control tumor growth has not met with the expected success. We suggest that this lack of success is because of AMPK-mediated energy metabolism rewiring, protecting cancer cell viability. On the basis of our observations, we propose an alternative therapeutic strategy of silencing either of the PKM isoforms along with AMPK in tumors.
Keywords: Warburg effect; cancer biology; cancer metabolism; cancer therapy; energy metabolism; mitochondria; mitochondrial oxidative phosphorylation; pyruvate kinase M1; pyruvate kinase M2.
© 2017 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
Similar articles
-
Apoptin Inhibits Glycolysis and Regulates Autophagy by Targeting Pyruvate Kinase M2 (PKM2) in Lung Cancer A549 Cells.Curr Cancer Drug Targets. 2024;24(4):411-424. doi: 10.2174/1568009623666221025150239. Curr Cancer Drug Targets. 2024. PMID: 36284386 Free PMC article.
-
Dietary-phytochemical mediated reversion of cancer-specific splicing inhibits Warburg effect in head and neck cancer.BMC Cancer. 2019 Nov 1;19(1):1031. doi: 10.1186/s12885-019-6257-1. BMC Cancer. 2019. PMID: 31675998 Free PMC article.
-
Knockdown of PKM2 induces apoptosis and autophagy in human A549 alveolar adenocarcinoma cells.Mol Med Rep. 2015 Sep;12(3):4358-4363. doi: 10.3892/mmr.2015.3943. Epub 2015 Jun 16. Mol Med Rep. 2015. PMID: 26082202
-
The Role of PKM2 in Metabolic Reprogramming: Insights into the Regulatory Roles of Non-Coding RNAs.Int J Mol Sci. 2021 Jan 25;22(3):1171. doi: 10.3390/ijms22031171. Int J Mol Sci. 2021. PMID: 33503959 Free PMC article. Review.
-
PKM2, cancer metabolism, and the road ahead.EMBO Rep. 2016 Dec;17(12):1721-1730. doi: 10.15252/embr.201643300. Epub 2016 Nov 17. EMBO Rep. 2016. PMID: 27856534 Free PMC article. Review.
Cited by
-
Inhibition of PTP1B blocks pancreatic cancer progression by targeting the PKM2/AMPK/mTOC1 pathway.Cell Death Dis. 2019 Nov 19;10(12):874. doi: 10.1038/s41419-019-2073-4. Cell Death Dis. 2019. PMID: 31745071 Free PMC article.
-
Intestinal epithelial PKM2 serves as a safeguard against experimental colitis via activating β-catenin signaling.Mucosal Immunol. 2019 Nov;12(6):1280-1290. doi: 10.1038/s41385-019-0197-6. Epub 2019 Aug 28. Mucosal Immunol. 2019. PMID: 31462699
-
Reprogramming of aerobic glycolysis in non-transformed mouse liver with pyruvate dehydrogenase complex deficiency.Physiol Rep. 2021 Jan;9(1):e14684. doi: 10.14814/phy2.14684. Physiol Rep. 2021. PMID: 33400855 Free PMC article.
-
The role of pyruvate kinase M2 in anticancer therapeutic treatments.Oncol Lett. 2019 Dec;18(6):5663-5672. doi: 10.3892/ol.2019.10948. Epub 2019 Oct 2. Oncol Lett. 2019. PMID: 31788038 Free PMC article. Review.
-
Identification of a six-gene metabolic signature predicting overall survival for patients with lung adenocarcinoma.PeerJ. 2020 Dec 2;8:e10320. doi: 10.7717/peerj.10320. eCollection 2020. PeerJ. 2020. PMID: 33344071 Free PMC article.
References
-
- Warburg O. (1956) On the origin of cancer cells. Science 123, 309–314 - PubMed
-
- Cairns R. A., Harris I. S., and Mak T. W. (2011) Regulation of cancer cell metabolism. Nat. Rev. Cancer 11, 85–95 - PubMed
-
- Christofk H. R., Vander Heiden M. G., Harris M. H., Ramanathan A., Gerszten R. E., Wei R., Fleming M. D., Schreiber S. L., and Cantley L. C. (2008) The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452, 230–233 - PubMed
-
- Mazurek S. (2011) Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell Biol. 43, 969–980 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
