

Experimental Modeling Supports a Role for MyBP-HL as a Novel Myofilament Component in Arrhythmia and Dilated Cardiomyopathy
- PMID: 28778945
- PMCID: PMC5645234
- DOI: 10.1161/CIRCULATIONAHA.117.028585
Experimental Modeling Supports a Role for MyBP-HL as a Novel Myofilament Component in Arrhythmia and Dilated Cardiomyopathy
Abstract
Background: Cardiomyopathy and arrhythmias are under significant genetic influence. Here, we studied a family with dilated cardiomyopathy and associated conduction system disease in whom prior clinical cardiac gene panel testing was unrevealing.
Methods: Whole-genome sequencing and induced pluripotent stem cells were used to examine a family with dilated cardiomyopathy and atrial and ventricular arrhythmias. We also characterized a mouse model with heterozygous and homozygous deletion of Mybphl.
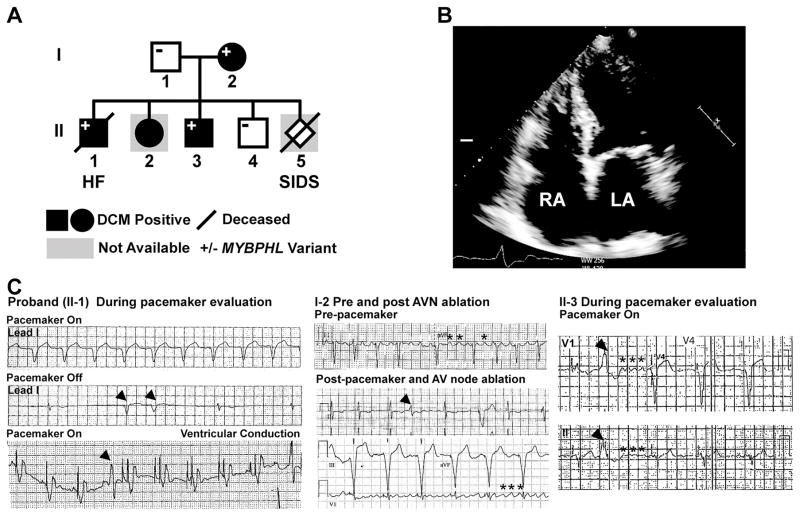
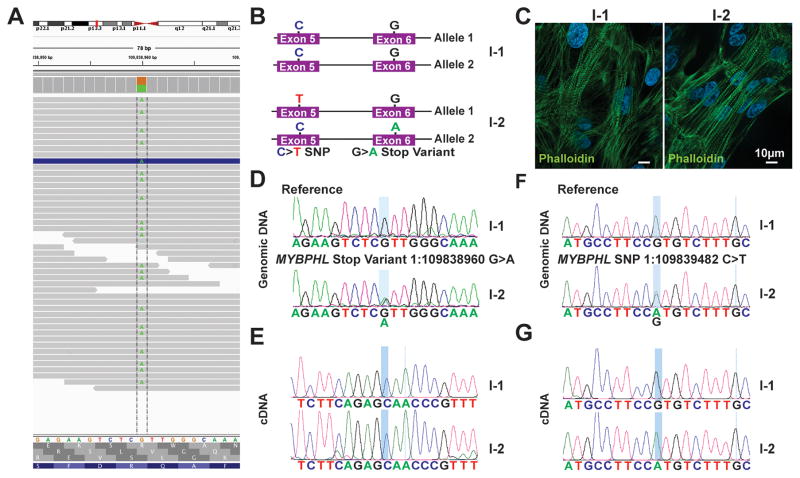
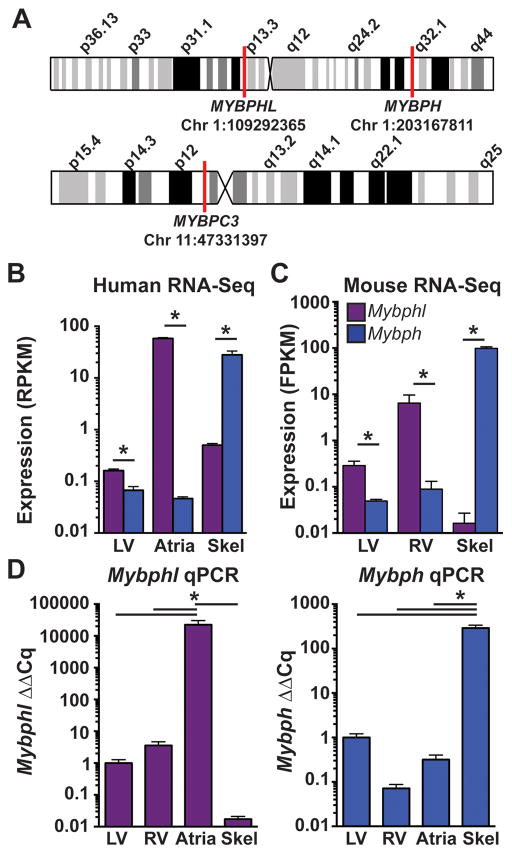
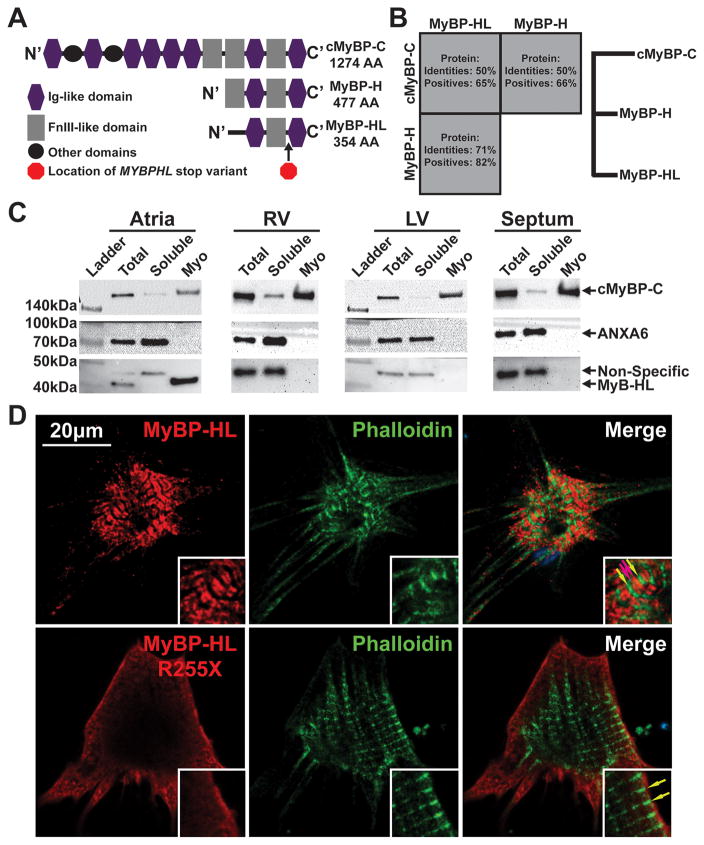
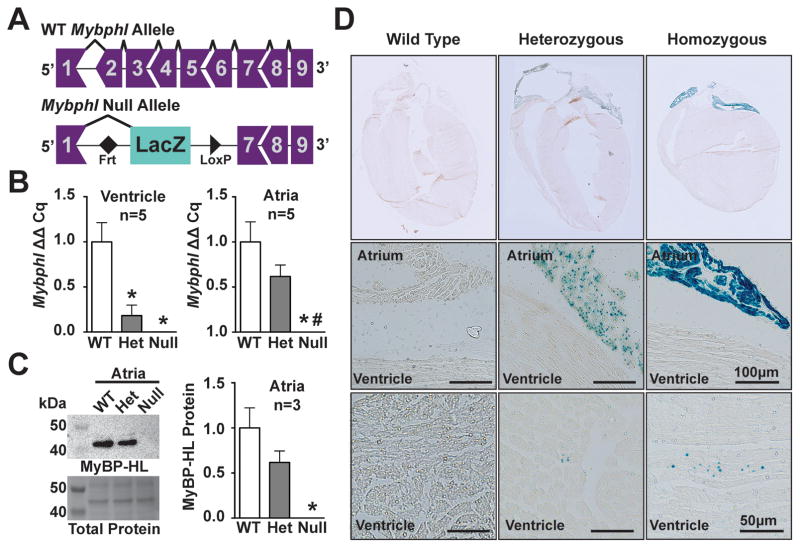
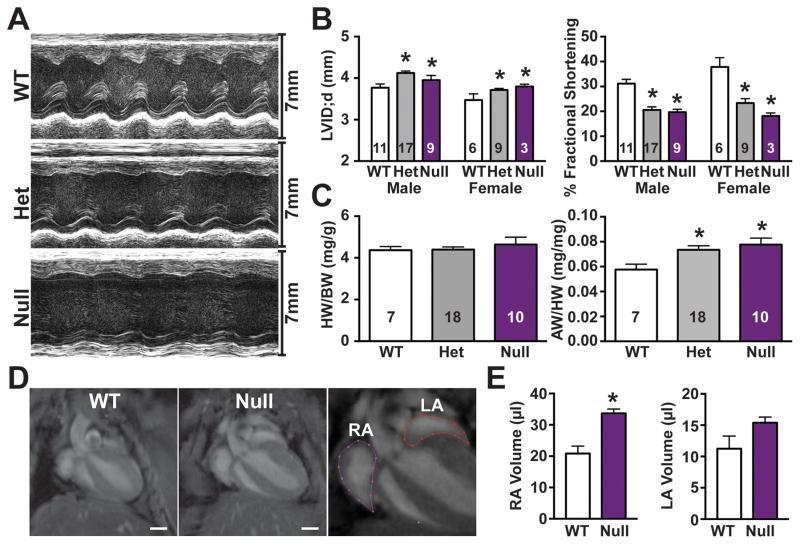
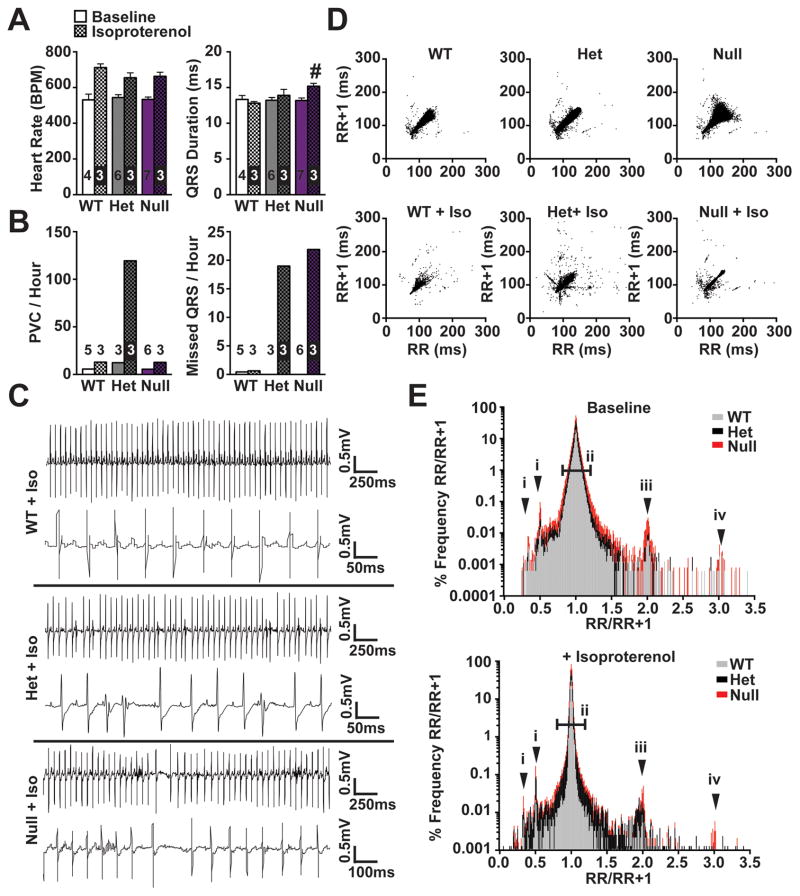
Results: Whole-genome sequencing identified a premature stop codon, R255X, in the MYBPHL gene encoding MyBP-HL (myosin-binding protein-H like), a novel member of the myosin-binding protein family. MYBPHL was found to have high atrial expression with low ventricular expression. We determined that MyBP-HL protein was myofilament associated in the atria, and truncated MyBP-HL protein failed to incorporate into the myofilament. Human cell modeling demonstrated reduced expression from the mutant MYBPHL allele. Echocardiography of Mybphl heterozygous and null mouse hearts exhibited a 36% reduction in fractional shortening and an increased diastolic ventricular chamber size. Atria weight normalized to total heart weight was significantly increased in Mybphl heterozygous and null mice. Using a reporter system, we detected robust expression of Mybphl in the atria, and in discrete puncta throughout the right ventricular wall and septum, as well. Telemetric electrocardiogram recordings in Mybphl mice revealed cardiac conduction system abnormalities with aberrant atrioventricular conduction and an increased rate of arrhythmia in heterozygous and null mice.
Conclusions: The findings of reduced ventricular function and conduction system defects in Mybphl mice support that MYBPHL truncations may increase risk for human arrhythmias and cardiomyopathy.
Keywords: arrhythmias, cardiac; cardiomyopathies; genetics; heart atria; mice; mutation; myofibrils.
© 2017 American Heart Association, Inc.
Conflict of interest statement
Figures
Comment in
-
Closing the Genotype-Phenotype Loop for Precision Medicine.Circulation. 2017 Oct 17;136(16):1492-1494. doi: 10.1161/CIRCULATIONAHA.117.030831. Circulation. 2017. PMID: 29038206 Free PMC article. No abstract available.
References
-
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB American Heart A, Council on Clinical Cardiology HF, Transplantation C, Quality of C, Outcomes R, Functional G, Translational Biology Interdisciplinary Working G, Council on E Prevention. Contemporary Definitions and Classification of the Cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287. - DOI - PubMed
-
- Lakdawala NK, Funke BH, Baxter S, Cirino AL, Roberts AE, Judge DP, Johnson N, Mendelsohn NJ, Morel C, Care M, Chung WK, Jones C, Psychogios A, Duffy E, Rehm HL, White E, Seidman JG, Seidman CE, Ho CY. Genetic Testing for Dilated Cardiomyopathy in Clinical Practice. J Card Fail. 2012;18:296–303. doi: 10.1016/j.cardfail.2012.01.013. - DOI - PMC - PubMed
-
- Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJ, Cook SA, Mestroni L, Seidman JG, Seidman CE. Truncations of Titin Causing Dilated Cardiomyopathy. N Engl J Med. 2012;366:619–628. doi: 10.1056/NEJMoa1110186. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
