

Shared ACVR1 mutations in FOP and DIPG: Opportunities and challenges in extending biological and clinical implications across rare diseases
- PMID: 28780023
- PMCID: PMC7888549
- DOI: 10.1016/j.bone.2017.08.001
Shared ACVR1 mutations in FOP and DIPG: Opportunities and challenges in extending biological and clinical implications across rare diseases
Abstract
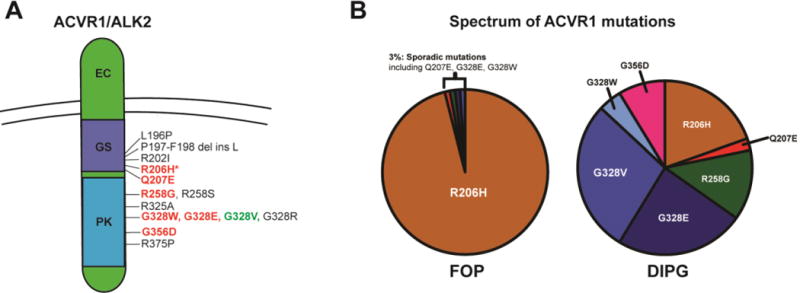
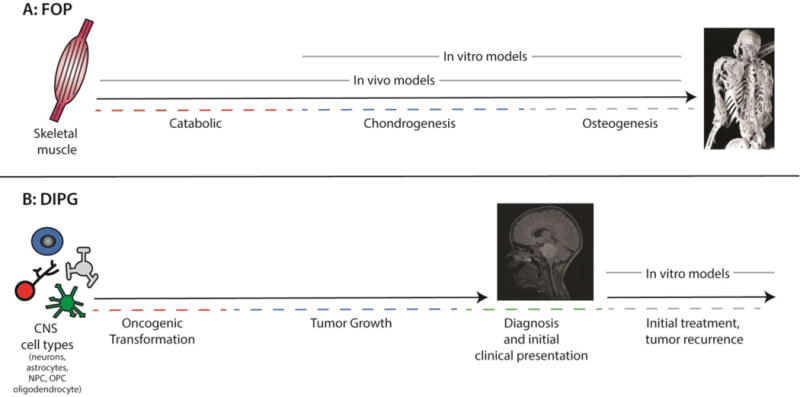
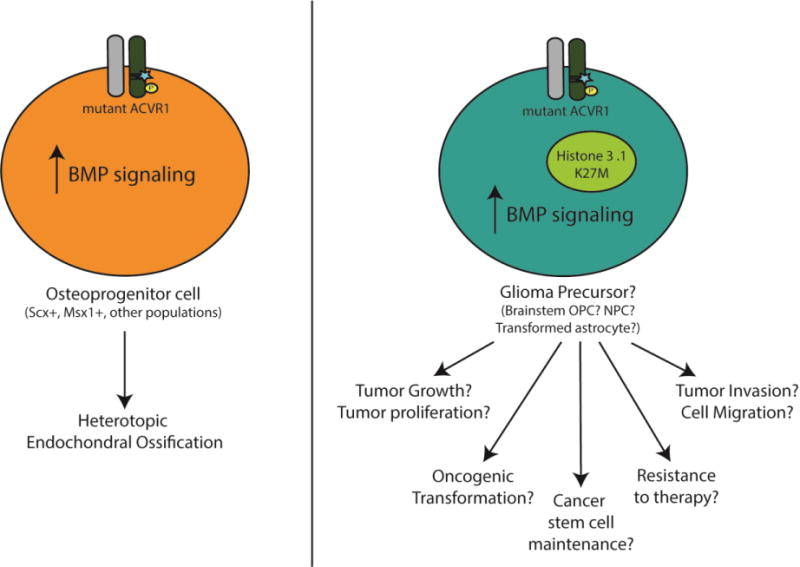
Gain-of-function mutations in the Type I Bone Morphogenic Protein (BMP) receptor ACVR1 have been identified in two diseases: Fibrodysplasia Ossificans Progressiva (FOP), a rare autosomal dominant disorder characterized by genetically driven heterotopic ossification, and in 20-25% of Diffuse Intrinsic Pontine Gliomas (DIPGs), a pediatric brain tumor with no effective therapies and dismal median survival. While the ACVR1 mutation is causal for FOP, its role in DIPG tumor biology remains under active investigation. Here, we discuss cross-fertilization between the FOP and DIPG fields, focusing on the biological mechanisms and principles gleaned from FOP that can be applied to DIPG biology. We highlight our current knowledge of ACVR1 in both diseases, and then describe the growing opportunities and barriers to effectively investigate ACVR1 in DIPG. Importantly, learning from other seemingly unrelated diseases harboring similar mutations may uncover novel mechanisms or processes for future investigation.
Keywords: ACVR1; BMP; Childhood cancers; Diffuse Intrinsic Pontine Glioma; Fibrodysplasia Ossificans Progressiva; Mendelian disorders.
Copyright © 2017 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Waite KA, Eng C. From developmental disorder to heritable cancer: it’s all in the BMP/TGF-beta family. Nat Rev Genet. 2003;4(10):763–73. - PubMed
-
- Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M, Morhart R, Rogers JG, Smith R, Triffitt JT, Urtizberea JA, Zasloff M, Brown MA, Kaplan FS. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet. 2006;38(5):525–7. - PubMed
-
- Kaplan FS, Xu M, Seemann P, Connor JM, Glaser DL, Carroll L, Delai P, Fastnacht-Urban E, Forman SJ, Gillessen-Kaesbach G, Hoover-Fong J, Koster B, Pauli RM, Reardon W, Zaidi SA, Zasloff M, Morhart R, Mundlos S, Groppe J, Shore EM. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum Mutat. 2009;30(3):379–90. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
