

Is there an optimum level of diversity in utilization of genetic resources?
- PMID: 28780586
- PMCID: PMC5641276
- DOI: 10.1007/s00122-017-2959-4
Is there an optimum level of diversity in utilization of genetic resources?
Abstract
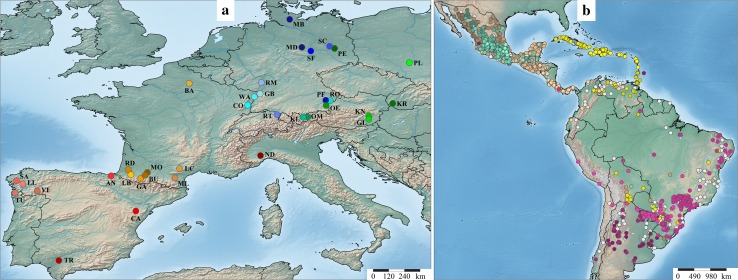
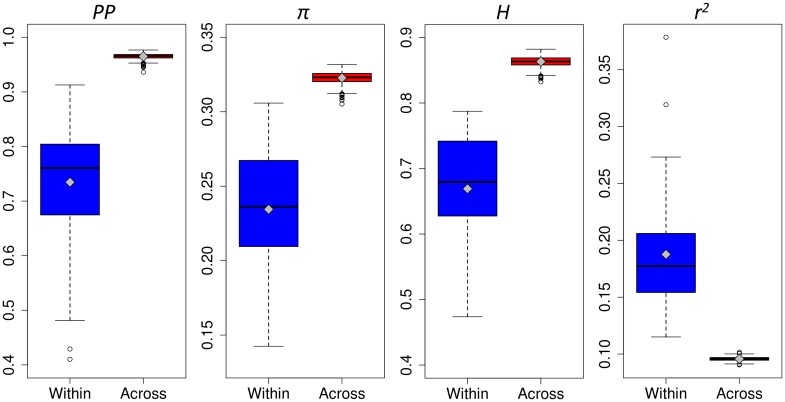
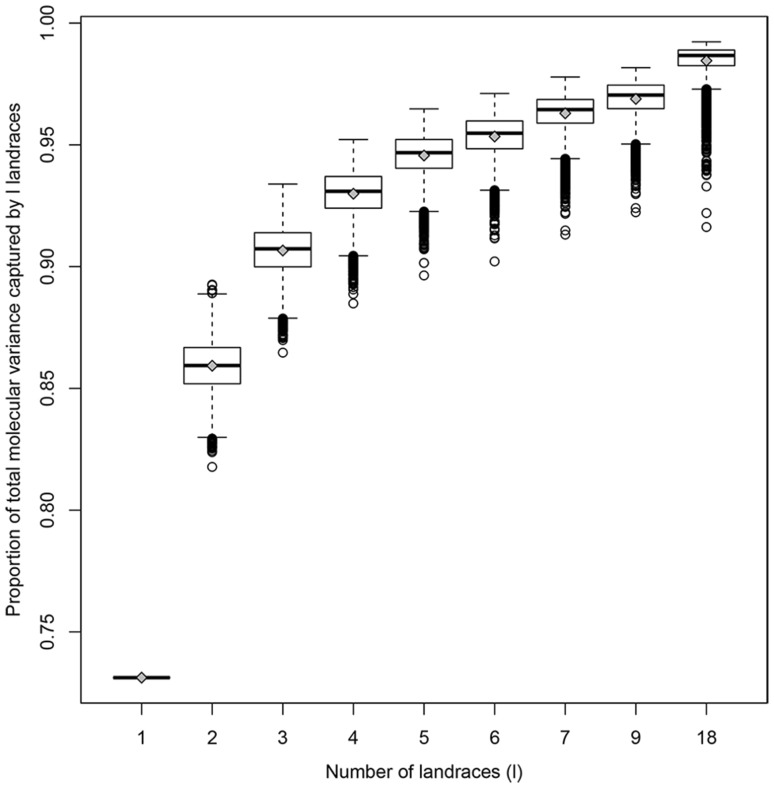
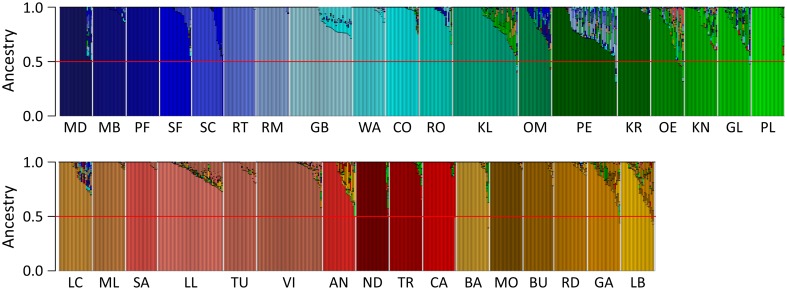
Capitalizing upon the genomic characteristics of long-term random mating populations, sampling from pre-selected landraces is a promising approach for broadening the genetic base of elite germplasm for quantitative traits. Genome-enabled strategies for harnessing untapped allelic variation of landraces are currently evolving. The success of such approaches depends on the choice of source material. Thus, the analysis of different strategies for sampling allelic variation from landraces and their impact on population diversity and linkage disequilibrium (LD) is required to ensure the efficient utilization of diversity. We investigated the impact of different sampling strategies on diversity parameters and LD based on high-density genotypic data of 35 European maize landraces each represented by more than 20 individuals. On average, five landraces already captured ~95% of the molecular diversity of the entire dataset. Within landraces, absence of pronounced population structure, consistency of linkage phases and moderate to low LD levels were found. When combining data of up to 10 landraces, LD decay distances decreased to a few kilobases. Genotyping 24 individuals per landrace with 5k SNPs was sufficient for obtaining representative estimates of diversity and LD levels to allow an informed pre-selection of landraces. Integrating results from European with Central and South American landraces revealed that European landraces represent a unique and diverse spectrum of allelic variation. Sampling strategies for harnessing allelic variation from landraces depend on the study objectives. If the focus lies on the improvement of elite germplasm for quantitative traits, we recommend sampling from pre-selected landraces, as it yields a wide range of diversity, allows optimal marker imputation, control for population structure and avoids the confounding effects of strong adaptive alleles.
Conflict of interest statement
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Ethical standards
The authors declare that this study complies with the current laws of the countries in which the experiments were performed.
Figures
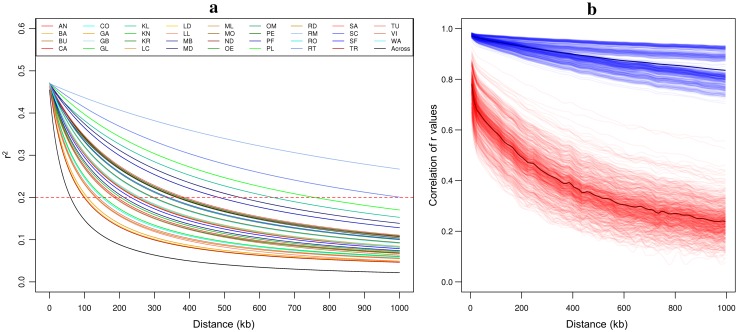
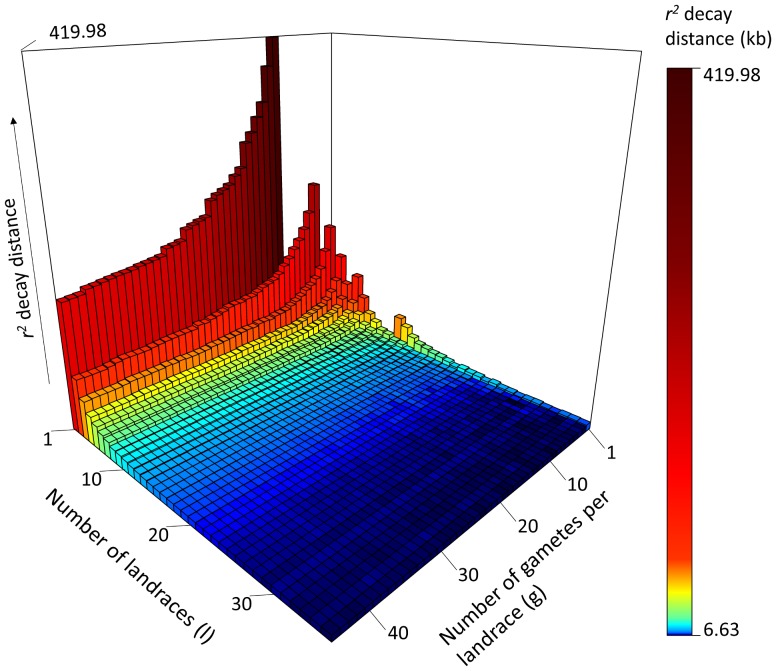
References
-
- Chia J-M, Song C, Bradbury PJ, Costich D, de Leon N, Doebley J, Elshire RJ, Gaut B, Geller L, Glaubitz JC, Gore M, Guill KE, Holland J, Hufford MB, Lai J, Li M, Liu X, Lu Y, McCombie R, Nelson R, Poland J, Prasanna BM, Pyhajarvi T, Rong T, Sekhon RS, Sun Q, Tenaillon MI, Tian F, Wang J, Xu X, Zhang Z, Kaeppler SM, Ross-Ibarra J, McMullen MD, Buckler ES, Zhang G, Xu Y, Ware D. Maize HapMap2 identifies extant variation from a genome in flux. Nat Genet. 2012;44:803–807. doi: 10.1038/ng.2313. - DOI - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
