

Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS
- PMID: 28783719
- PMCID: PMC5648058
- DOI: 10.1038/nature23291
Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS
Abstract
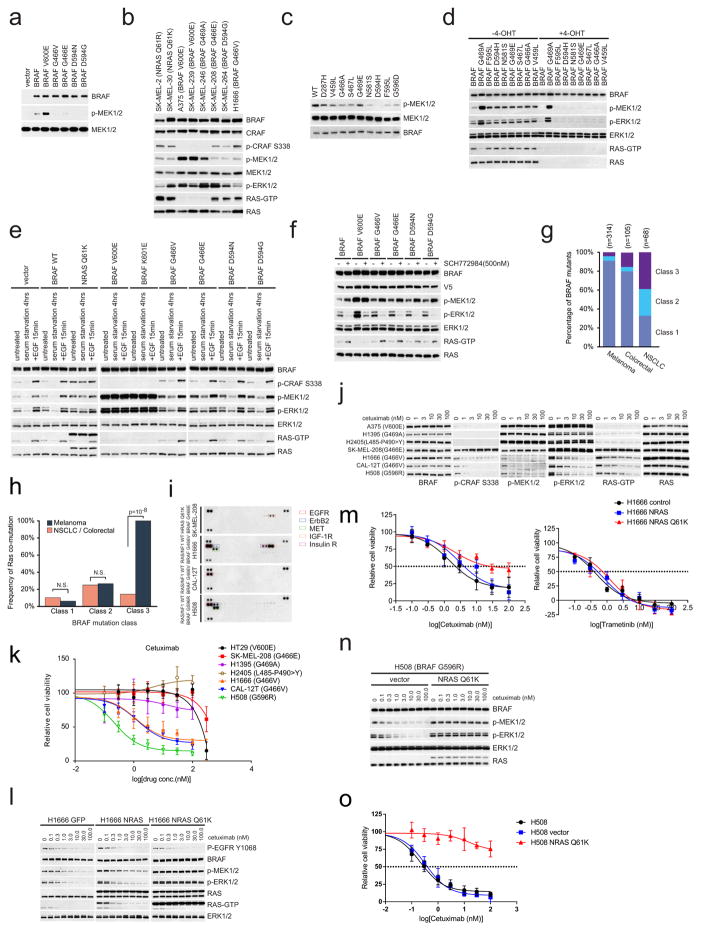
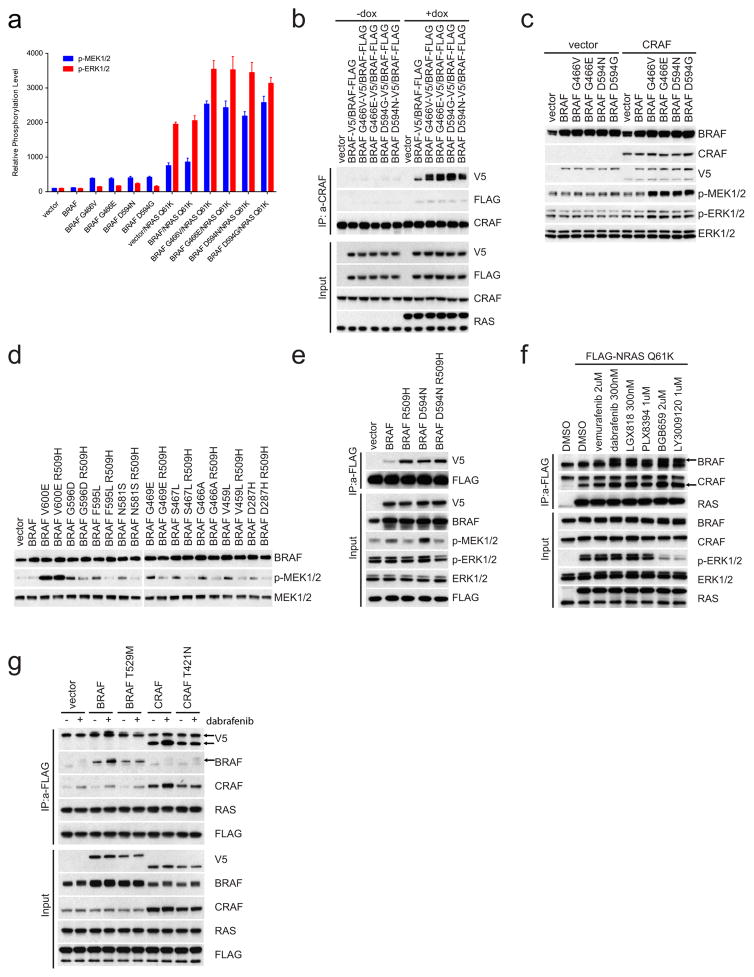
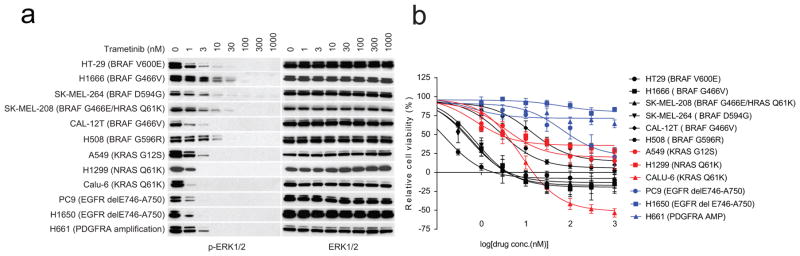
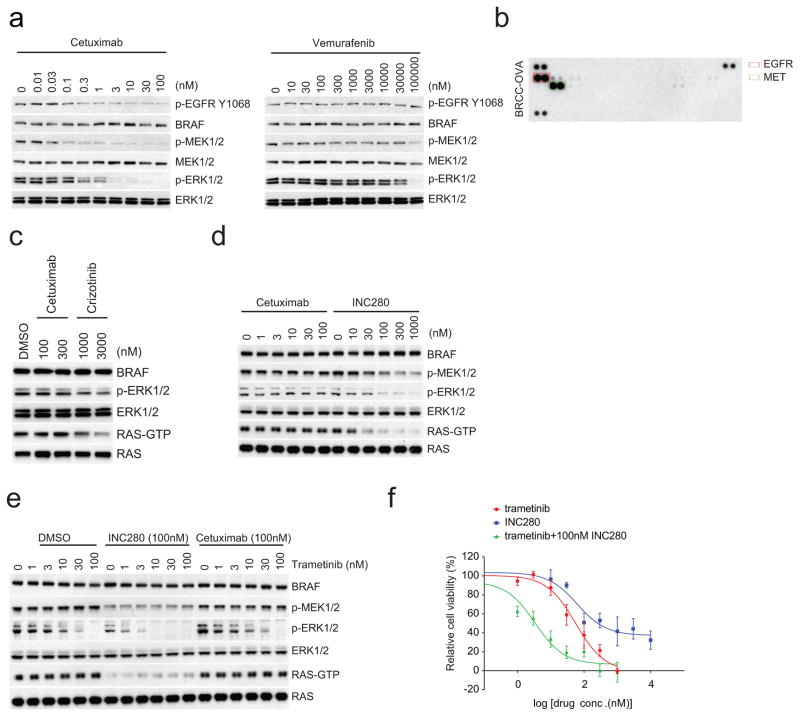
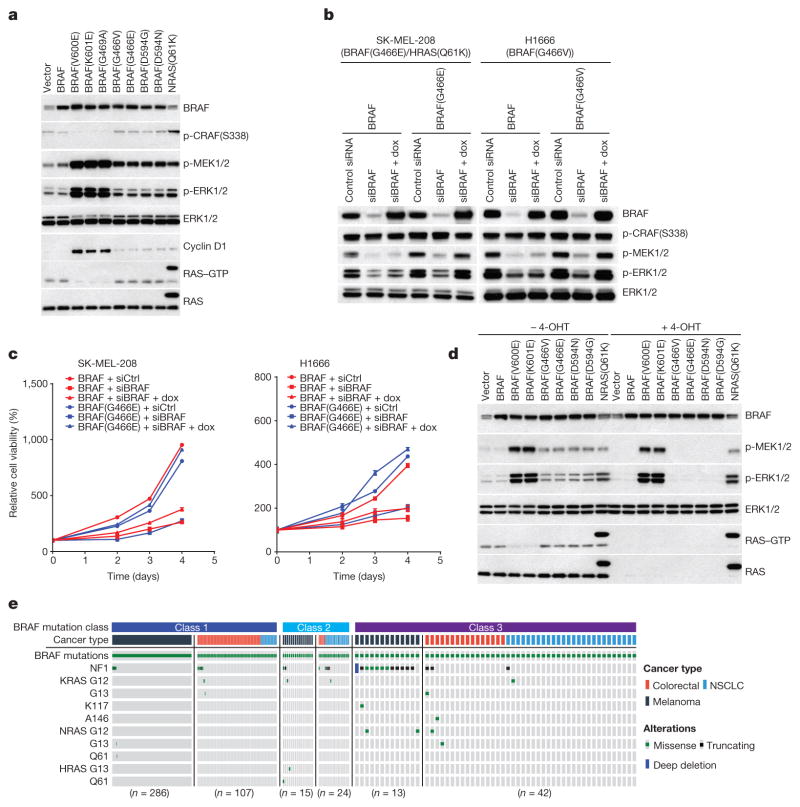
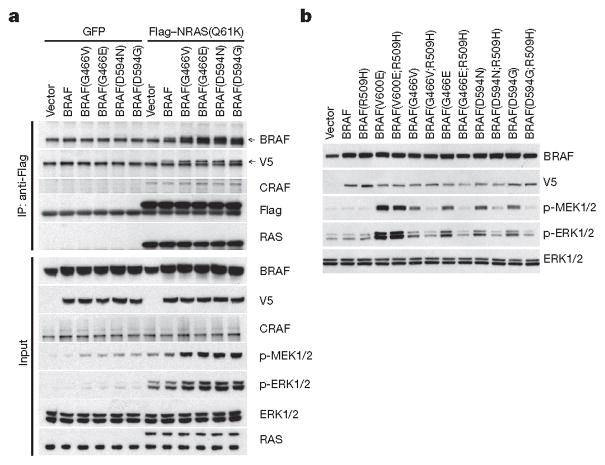
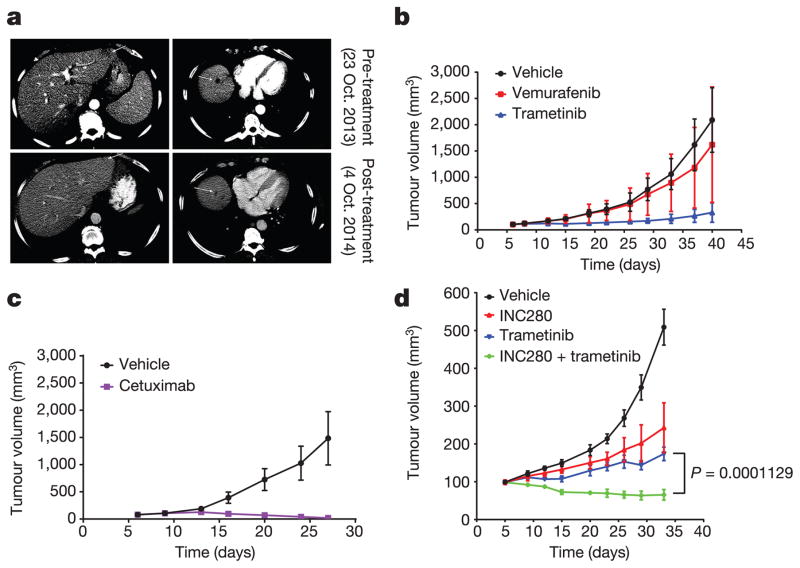
Approximately 200 BRAF mutant alleles have been identified in human tumours. Activating BRAF mutants cause feedback inhibition of GTP-bound RAS, are RAS-independent and signal either as active monomers (class 1) or constitutively active dimers (class 2). Here we characterize a third class of BRAF mutants-those that have impaired kinase activity or are kinase-dead. These mutants are sensitive to ERK-mediated feedback and their activation of signalling is RAS-dependent. The mutants bind more tightly than wild-type BRAF to RAS-GTP, and their binding to and activation of wild-type CRAF is enhanced, leading to increased ERK signalling. The model suggests that dysregulation of signalling by these mutants in tumours requires coexistent mechanisms for maintaining RAS activation despite ERK-dependent feedback. Consistent with this hypothesis, melanomas with these class 3 BRAF mutations also harbour RAS mutations or NF1 deletions. By contrast, in lung and colorectal cancers with class 3 BRAF mutants, RAS is typically activated by receptor tyrosine kinase signalling. These tumours are sensitive to the inhibition of RAS activation by inhibitors of receptor tyrosine kinases. We have thus defined three distinct functional classes of BRAF mutants in human tumours. The mutants activate ERK signalling by different mechanisms that dictate their sensitivity to therapeutic inhibitors of the pathway.
Conflict of interest statement
The authors declare competing financial interests: details are available in the online version of the paper.
Figures

Comment in
-
Cell signalling: Even kinase-inactive BRAF is oncogenic.Nat Rev Clin Oncol. 2017 Nov;14(11):648-649. doi: 10.1038/nrclinonc.2017.140. Epub 2017 Aug 31. Nat Rev Clin Oncol. 2017. PMID: 28857078 No abstract available.
References
-
- Wan PT, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
