

Evaluation and comparison of computational tools for RNA-seq isoform quantification
- PMID: 28784092
- PMCID: PMC5547501
- DOI: 10.1186/s12864-017-4002-1
Evaluation and comparison of computational tools for RNA-seq isoform quantification
Abstract
Background: Alternatively spliced transcript isoforms are commonly observed in higher eukaryotes. The expression levels of these isoforms are key for understanding normal functions in healthy tissues and the progression of disease states. However, accurate quantification of expression at the transcript level is limited with current RNA-seq technologies because of, for example, limited read length and the cost of deep sequencing.
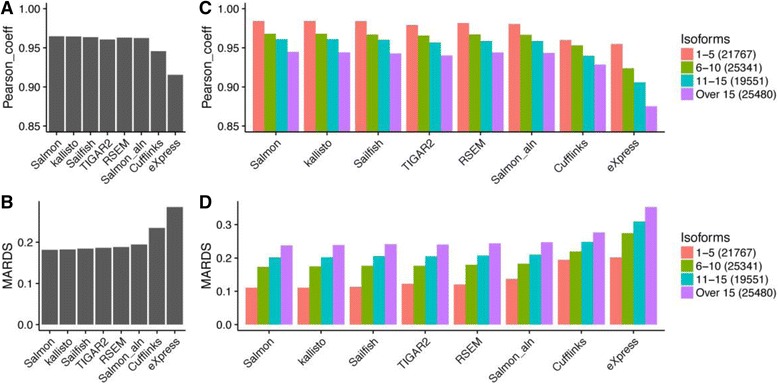
Results: A large number of tools have been developed to tackle this problem, and we performed a comprehensive evaluation of these tools using both experimental and simulated RNA-seq datasets. We found that recently developed alignment-free tools are both fast and accurate. The accuracy of all methods was mainly influenced by the complexity of gene structures and caution must be taken when interpreting quantification results for short transcripts. Using TP53 gene simulation, we discovered that both sequencing depth and the relative abundance of different isoforms affect quantification accuracy CONCLUSIONS: Our comprehensive evaluation helps data analysts to make informed choice when selecting computational tools for isoform quantification.
Keywords: Data analysis; Isoform; Kallisto; Quantification; RNA-seq; RSEM; Salfish; Salmon.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures






References
-
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–515. doi: 10.1038/nbt.1621. - DOI - PMC - PubMed
-
- Batra R, Charizanis K, Manchanda M, Mohan A, Li M, Finn DJ, Goodwin M, Zhang C, Sobczak K, Thornton CA, et al. Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol Cell. 2014;56(2):311–322. doi: 10.1016/j.molcel.2014.08.027. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
