

Cardiotrophin 1 stimulates beneficial myogenic and vascular remodeling of the heart
- PMID: 28785017
- PMCID: PMC5630684
- DOI: 10.1038/cr.2017.87
Cardiotrophin 1 stimulates beneficial myogenic and vascular remodeling of the heart
Abstract
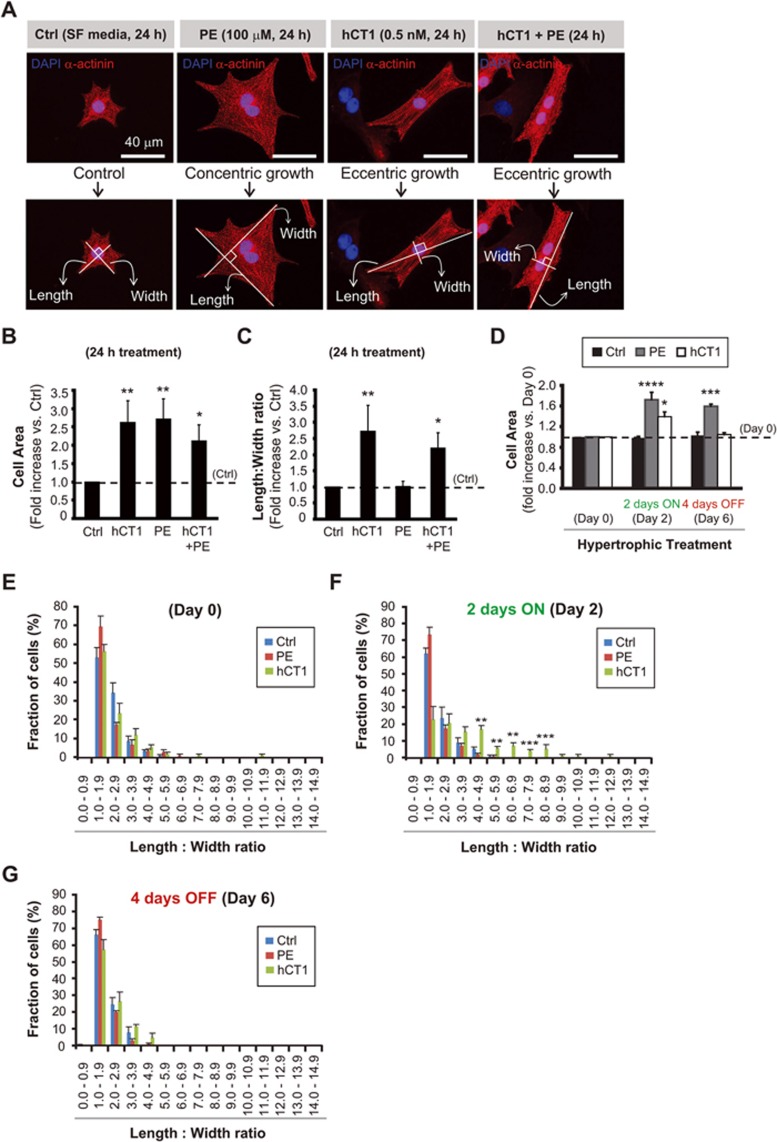
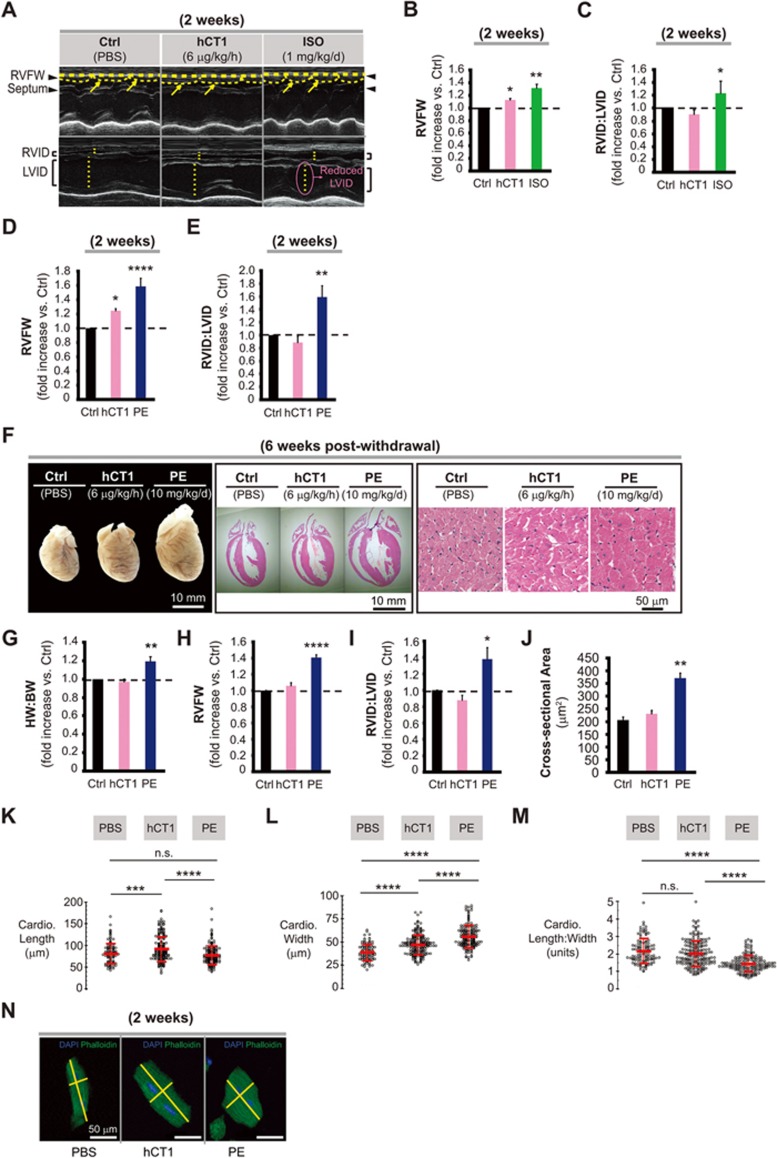
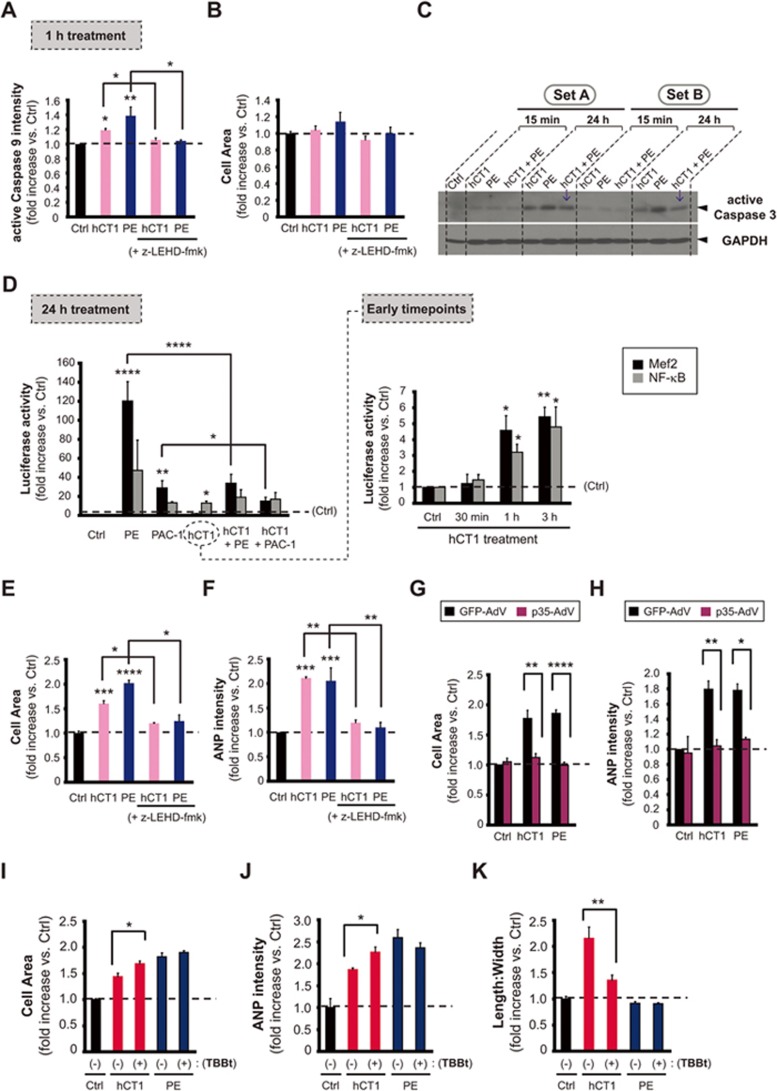
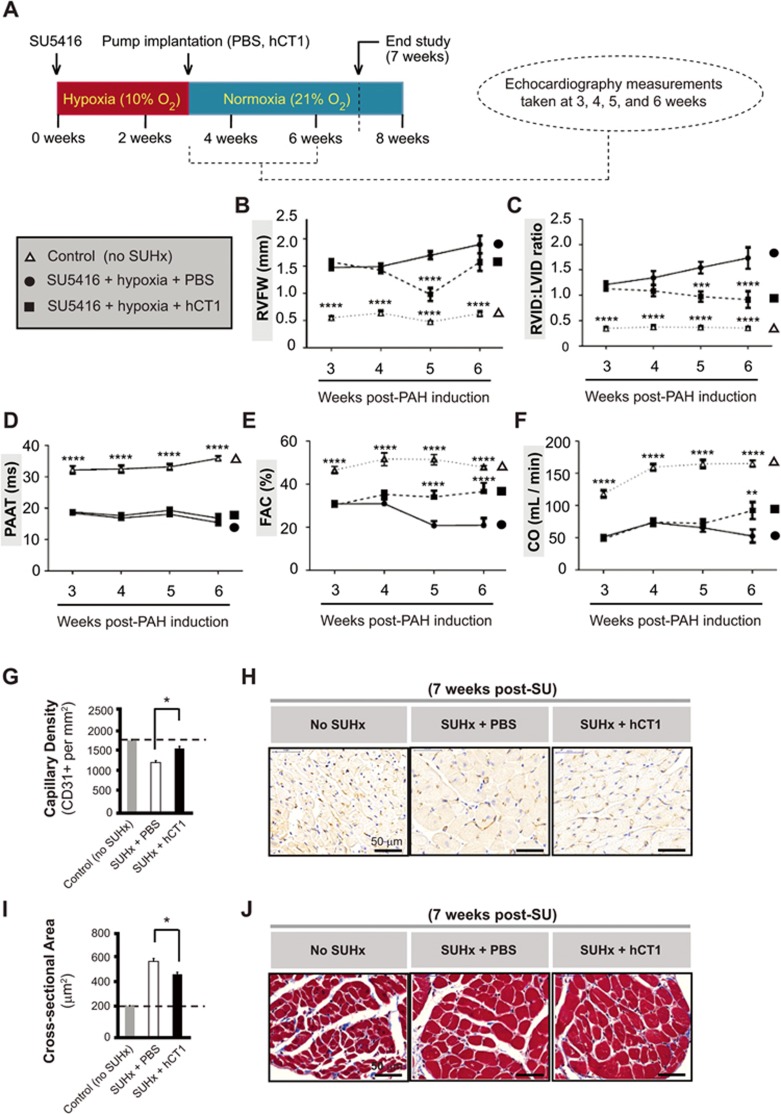
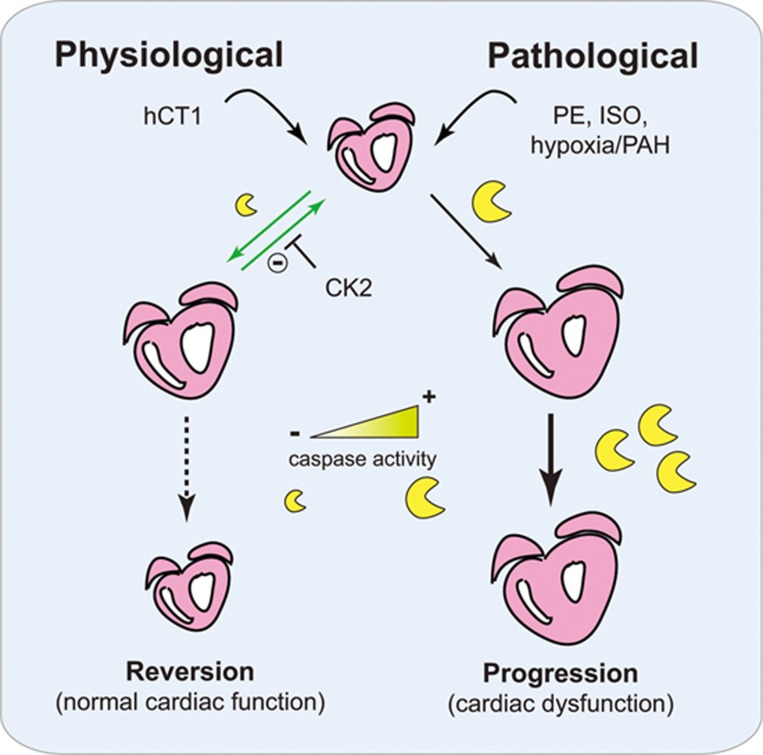
The post-natal heart adapts to stress and overload through hypertrophic growth, a process that may be pathologic or beneficial (physiologic hypertrophy). Physiologic hypertrophy improves cardiac performance in both healthy and diseased individuals, yet the mechanisms that propagate this favorable adaptation remain poorly defined. We identify the cytokine cardiotrophin 1 (CT1) as a factor capable of recapitulating the key features of physiologic growth of the heart including transient and reversible hypertrophy of the myocardium, and stimulation of cardiomyocyte-derived angiogenic signals leading to increased vascularity. The capacity of CT1 to induce physiologic hypertrophy originates from a CK2-mediated restraining of caspase activation, preventing the transition to unrestrained pathologic growth. Exogenous CT1 protein delivery attenuated pathology and restored contractile function in a severe model of right heart failure, suggesting a novel treatment option for this intractable cardiac disease.
Figures

Comment in
-
Size matters: Finding growth pathways that protect the heart.Cell Res. 2017 Oct;27(10):1187-1188. doi: 10.1038/cr.2017.120. Epub 2017 Sep 19. Cell Res. 2017. PMID: 28925384 Free PMC article.
References
-
- Hill JA, Olson EN. Cardiac plasticity. N Engl J Med 2008; 358:1370–1380. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
